




The heparin debacle and other crises involving imported drugs and biologics has put pressure on the US FDA to step up its oversight of foreign drug manufacturing.

The country is reeling from US Treasury Secretary Henry Paulson's proposed $700 billion Wall Street bailout. In appealing for support, Paulson stressed this was a crisis. This sense of urgency is reminiscent of the way millions of Americans without health insurance cope with medical problems-in the emergency room, because they have no access to routine and preventive care.
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
The heparin debacle and other crises involving imported drugs and biologics has put pressure on the US FDA to step up its oversight of foreign drug manufacturing.

Risk mitigation should be a key aspect of any contract manufacturing organization's business strategy.

The complexity of Quality by Design leads naturally to questions of how much work it requires, how many companies have the resources to do it, and what the payoff is for anybody.

GMPs in Phase 1 is more important now than ever before.
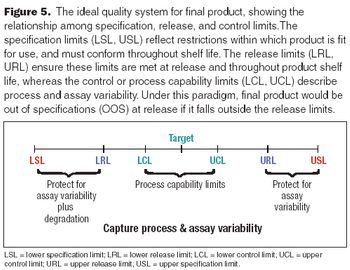
Set limits to provide incentives for process improvements.

Manufacturers of biopharmaceuticals can improve productivity by taking patient wellness into account.
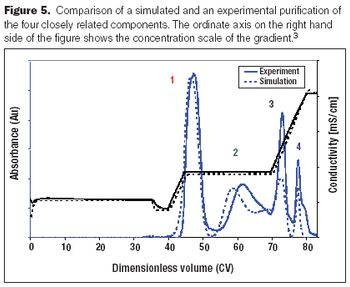
A review of some recent contributions in process chromatography.

The Sentinel System aims to generate more adverse event reporting by health professionals, to analyze health information more effectively, and to enhance FDA methods for communicating new safety information to providers and patients.
Enough Republicans sided with Democrats last month to approve legislation canceling a scheduled 10.6% cut in Medicare fees for physicians. In doing so, the legislators tacked on dozens of provisions pleasing to beneficiaries and providers alike.
The FDA is launching a pilot program to allow manufacturers to electronically file drug establishment registration and drug listing information, such as ingredients, labeling, and manufacturing information.
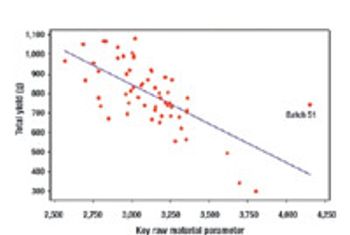
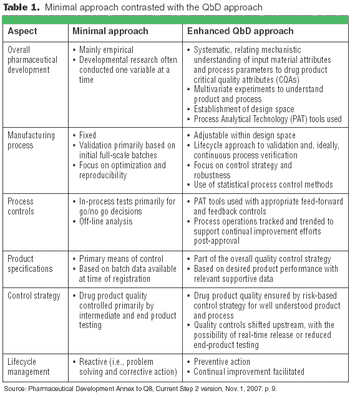
Quality by Design and Design Space can be used by companies to enhance process understanding, improve scientific rigor, and enhanced qualitative and quantative performance, as well as cost savings.

The US Food and Drugs Administration is boosting its efforts for orphan drugs development.

Best practices a pharmaceutical company can follow to execute their strategy sucessfully.
Resolve confusion about measurements.

Pushing frontiers in a maturing biotechnology industry.
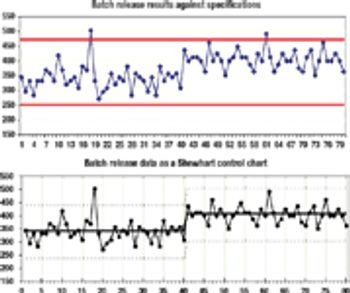
Improving your quality operations by using sound statistical principles.

How to use hypothesis correctly, and understanding the difference between one-sample, two-sample, and z-test.

It is important to understand critical aspects of the CMO's capabilities. Only by auditing certain key areas can the sponsor be assured of the quality of the materials produced.

The comparative research approach may be preferable to price controls in the guise of government negotiations for the Medicare drug benefit, coverage denials, and limits on access to new technologies.

A staged approach to limits should embrace future capabilities.

The industry needs to open up to validation failures.
US Food and Drug Administration's Division of Biologic Oncology Products has approved two new biologics license application (BLA) supplements expanding the approval of Genentech's Herceptin (trastuzumab) for the treatment of breast cancer.

The FDA is under attack from all sides. Many influential members of Congress either don't trust the agency to monitor the industry appropriately, or have found it politically expedient to keep sounding alarms about inadequate oversight of food and drug safety and clinical research. The good news is that there seems to be a growing consensus that FDA needs a major infusion of cash to regain its stature as an effective science-based regulatory agency.
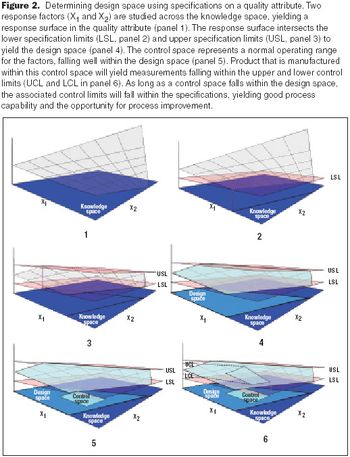
The principles of QbD can be applied to biotech development and manufacturing to help resolve many common issues. QbD scientifically provides a greater understanding of the complex relationships among product quality attributes, the manufacturing process, and clinical safety and efficacy by determining the various permutations of critical input variables that will keep the product within specification.
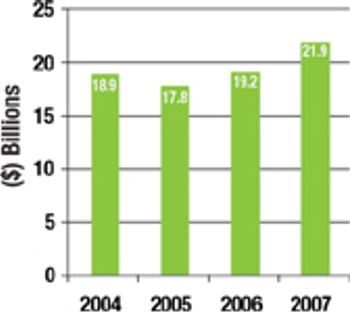
The current overcapacity situation in the bio/pharmaceutical industry is a reminder that CMOs need to come up with business models and value propositions that are based on more than just selling capacity.