

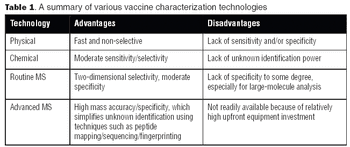
With the advent of high-resolution mass spectrometers and highly sensitive MS instruments, vaccine characterization has entered a new phase.
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
With the advent of high-resolution mass spectrometers and highly sensitive MS instruments, vaccine characterization has entered a new phase.

Any endpoint considered appropriate to support approval, whether a surrogate or a clinical endpoint, must be supported by substantial evidence of effectiveness.
The corruption of China's food and drug sectors is not limited to one man. Likewise, real reform will require the efforts of many.

Understanding the end-to-end management of chemistry, manufacturing, and controls (CMC) resources provides the opportunity to enhance long-term planning, leverage development options, manage resource trade offs, and track progress against plans. The goal is to improve the pharmaceutical development process to deliver the pipeline. This article provides an overview of the organizational structure of Process Research and Development (PR&D) and the CMC teams at Genentech; the alignment of resources based on CMC contracts, process development activity maps and project resource plans; and the business economic analysis for evaluating development options.

The main testing and regulatory provisions of the FOB legislation reflect multiple trade-offs between the demands of innovators and generics firms.
The US Pharmacopeia (Rockville, MD, USP, www.usp.org) recently announced that the implementation period for its USP–NF general notices statement requiring all manufacturers to conform to recently revised residual solvent standards in General Chapter <467> has been extended from July 1, 2007 to July 1, 2008.
The US FDA (Rockville, MD, www.fda.gov) recently announced the availability of a draft guidance, entitled, Q10 Pharmaceutical Quality System.
In late June and early July, the US Congress moved forward on three important bills affecting the biopharmaceutical industry, related to follow-on biologics, the Prescription Drug User Fee Act (PDUFA), and the 2008 FDA budget.

In the April issue of BioPharm International, the article "BioPharmaceutical Operations Roadmap," provided a summary of the key industry gaps executives would like to close in the next 10 years. These goals came to my mind while I was attending Interphex this April in New York.

Now equipment makers are designing new systems that can be deployed rapidly, are less costly to build, and require less water and power to operate.

Quality guidelines are only as good as their implementation.

The FDA issued MedImmune, Inc. (Gaithersburg, MD, www.medimmune.com), a warning letter for violating the agency's manufacturing rules and held off approving the company's influenza vaccine for use in children younger than age five until the problems are resolved.
The inaugural West Coast meeting for the Clinical Supplies Support Group (CSSG, www.jeiven.com/clinical_supplies_support_group.htm) was held in San Diego on June 8, 2007.
The US Food and Drug Administration (FDA, Rockville, MD, www.fda.gov) has issued final recommendations for increasing the supply of safe and effective influenza vaccines for both seasonal and pandemic use.
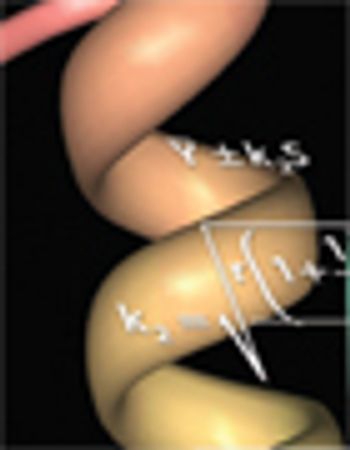
One goal of process characterization is establishing representative performance parameter ranges that can be used to set validation acceptance criteria (VAC). Characterization studies yield varying numbers of data points from multiple experiments, and may also include data generated at different scales (e.g., bench, pilot, and commercial), which add complexity to the analysis. Many statistical approaches can be used to set ranges from large data sets. As an example, we present the statistical considerations and techniques for setting validation acceptance ranges for a chromatography step used in purifying a recombinant protein. Performance parameter data from a combined data set consisting of 67 bench, six pilot, and three full-scale runs were analyzed using the statistical analysis software JMP (SAS Institute). The combined data set was used to compute tolerance intervals, so that sources such as scale and column feed material could be properly modeled. The resulting ranges were used to establish..

Outsourcing has been a cornerstone of our industry for decades.
FDA's approach involves adopting efficient strategies for targeting inspections to more high-risk operations likely to have the greatest impact on public health.
Following the US Senate approval of the FDA Revitalization Act (FDARA, S.1082) in May, the debate over drug safety and the reauthorization of the Prescription Drug User Fee Act (PDUFA) now moves to the House.

Membrane-based chromatography technologies sometimes offer advantages over resin-based technologies.

The information provided by analytical testing is important in determining whether additional clinical trails are necessary to bring a follow-on to market.

Lax enforcement arising from a lack of political will creates the potential for a loss of public confidence.
In March, the US Food and Drug Administration released a new draft guidance, along with its guidance agenda for the year.
Neotropix, Inc. (Malvern, PA, www.neotropix.com), a biotechnology company dedicated to the development and commercialization of virus-based therapeutics for the treatment of cancer and other diseases, received a warning letter (http://www.fda.gov/foi/warning_letters/b6308d.pdf) on March 23, 2007, citing deviations from good laboratory practices (GLP) regulations governing the proper conduct of nonclinical studies as published under 21 CFR Part 58.

A tremendous amount of analytical testing is required to support a biopharmaceutical product from discovery, development, and clinical trials, through manufacturing and marketing. Numerous methods are used to fully characterize large molecules because of their complexity-characterizing them is significantly more difficult than it is for small molecules. Biopharmaceuticals are produced via living systems, i.e., E. coli, yeast, or mammalian cells, which require additional testing matrices.