After a strategic evaluation of what activities to outsource, sponsor companies should follow key guidelines for selecting and auditing a provider and preparing quality agreements.
Quality/GMPs
Latest News
Advertisement
Digitalization of QbD Risk Assessments
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

Subjectivity in Quality Risk Management
The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Phase-appropriate Compliance for Cell and Gene Therapies
Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
Advertisement
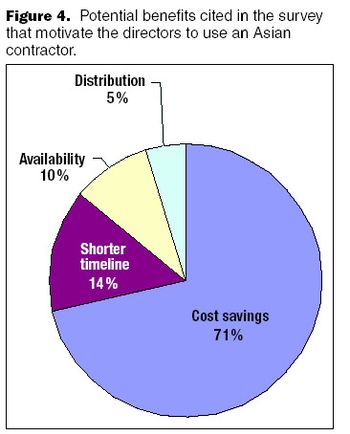
When a biopharmaceutical company pursues an outsourcing strategy, the choice of a contractor is a critical and strategic decision.

The book is a useful, comprehensive, and truly an excellent reference source of biopharmaceutical information.

Engaging executive leadership in the quality process is the key to compliance success.

A case study investigated the root cause of failures in sterile filtration by evaluating the effects and interactions of four operating parameters.

Did the 2005 Patents Act engender a Western intellectual property rights culture in the country?

The heparin safety crisis puts a spotlight on manufacturing processes and regulatory oversight.

Now is a good time for companies to know their suppliers well.

The supply base for preclinical and clinical development services continues to expand in China.

India is restructuring its regulation of biopharmaceuticals to help the country's industry compete internationally.

More informed submissions may lead to regulatory flexibility for postapproval changes.
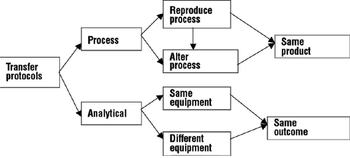
A comprehensive process and analytical transfer package can speed up your product's time to market and save costs.
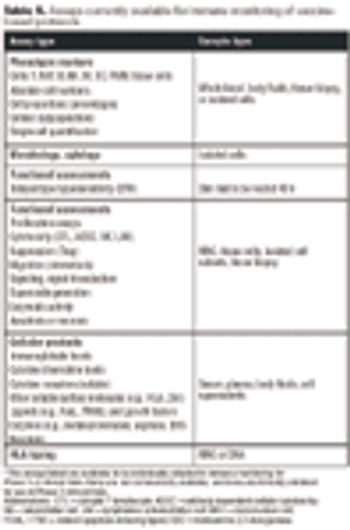
Emerging therapies pose challenges for standardizing QC.

The expanding globalization of the industry poses a challenge for international enforcement.
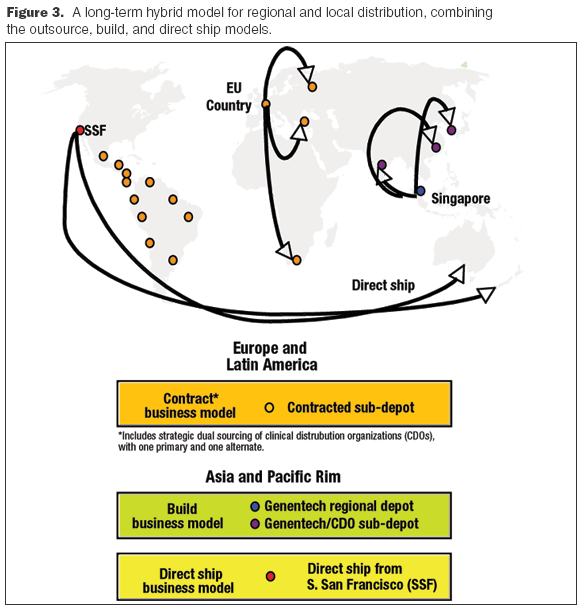
Need a standard to follow? Just ask, What would Genentech do?
Following the partial recall initiated on January 25, 2008, Baxter Healthcare has expanded its recall of heparin.
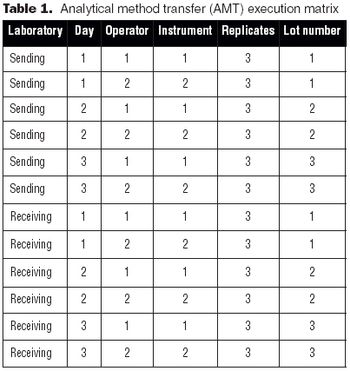
It is essential to understand the critical elements of validation extensions to ensure accurate process or product quality measurements.
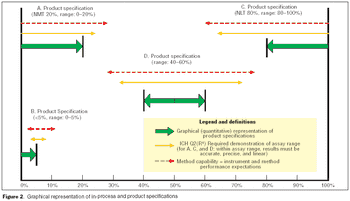
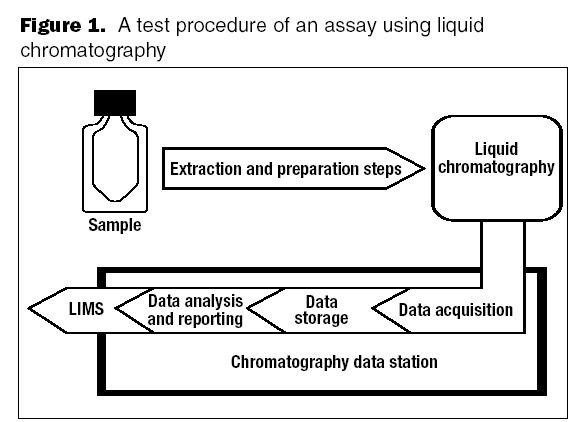
Validation of analytical methods can be more easily accomplished by breaking the task down into a series of planned steps.
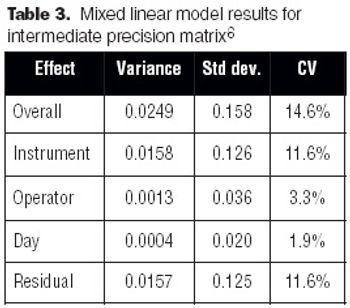
The development and optimization process can improve a method, but validation does not. Validation is the final proof that regulations and expectations are met.
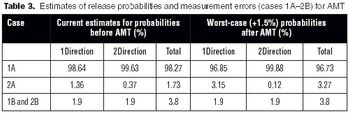
Several steps can be taken to maintain test method suitability after the formal completion of the analytical method validation studies,

Two case studies illustrate a systematic approach.

The much needed modernization of the agency's IT systems and inspection capabilities will likely fall prey to budget shortfalls.

Both innovator and generics companies are using analytics to support comparability arguments.
Best practices from Big Biotech, including how to handle new product introductions.
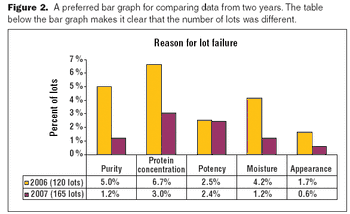
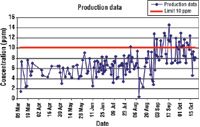
The key to a good graphical presentation is to select the method that best fits the data.
Advertisement
Advertisement