


While not yet finalized and adopted, ICH Q10 represents some of the most current thinking with respect to pharmaceuticals manufacturing and control.


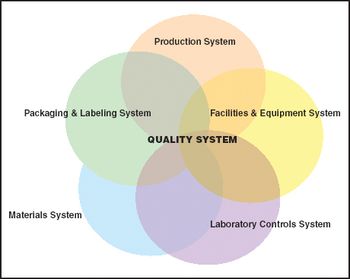
From the Editor: We All Need Quality Systems Even at the Cellular Level
While not yet finalized and adopted, ICH Q10 represents some of the most current thinking with respect to pharmaceuticals manufacturing and control.

Development reports document process development and support the design of validation experiments, yet in many firms training is not provided nor are expectations established. This article describes how project managers can help scientists master the art of report-writing.

SOPs are written job aids that detail the procedure of how to do a specific job task correctly.

The concept of design space has started a minor revolution in our industry.

Reserve samples of test and control articles must be retained for at least one stability time point after the completion of the study.

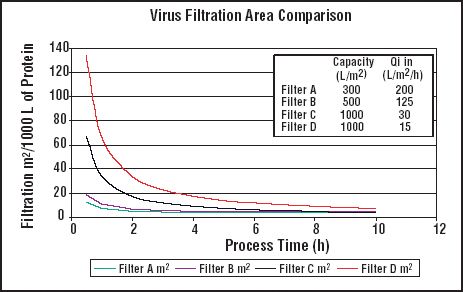
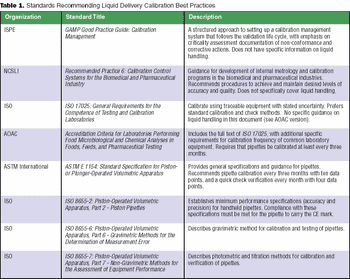
The challenge is to determine the optimal frequency for preventive maintenance and the optimal frequency and tolerances for calibration readings.

Taking a purist stance is always tempting. It can, however, have unintended consequences.
Companies faced with real or threatened FDA sanctions are usually least prepared to react effectively.

Increasingly, pharmaceutical companies have recognized that software development is not their core competency.

Good risk management tools dictate how much assay performance characteristics can deviate from ideal.

There is growing support for new partnership arrangements that seek to expand drug research by reducing the financial risk for manufacturers.

Understand your company's requirements, define responsibilities,and manage your team effectively.

Is it possible to reconcile phage therapy, which is inherently variable, with requirements for tight product characterization and control?

FDA did not gain any real teeth for regulating unsafe and ineffective products until a national health disaster in 1937 roused a public outcry.

In 2005, 10 biopharma- ceuticals gained marketing approval in the US or Europe, although only five of them were genuinely new molecular entities.
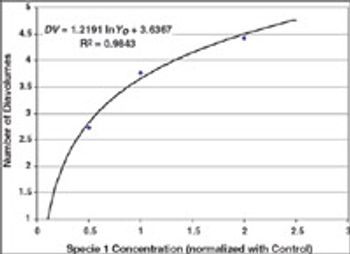
Case studies were run to test Process Analytical Technology applications for protein refolding, diafiltration, and cation exchange chromatography. It is shown that it is feasible to design control schemes that rely on measurement of product quality attributes and thereby enable real-time decisions.
Federal regulations are broad and open to interpretation. Most have not caught up with advancements in technology.

Disposables require less space than conventional equipment, and they can be assembled offsite into complete process trains.

A novel calibration approach was developed that not only calibrates the X-axis, but also calibrates the peak shape.

For decades now, it has been said that "the process is the product" for biologics. Great care and consistency must be applied in their upstream manufacture-during fermentation, harvest, and early purification-to preserve their complex structure, which confers their activity and specificity. As the product moves to late-stage purification, however, the relative concentration of impurities and altered product forms is diminished. Also, the final dosage form of most large molecule biopharmaceuticals is the relatively simple liquid formulation of parenteral dosage form. In contrast, manufacturing the solid dosage forms common for small-molecule drugs involves more complex processes, such as mixing dry powders, granulation, manufacturing controlled-release matrices, and tableting.

Food and Drug Administration is encouraging public–private collaborations to more fully explore the physical and chemical characteristics of nanoparticles.
A symposium at the AAPS National Biotechnology Conference in Boston, June 19-22, 2006, addressed key concerns and new developments in manufacturing biologic products in a sterile environment.
"Clinical data is the gold standard" for setting manufacturing specifications, said Patrick Swann, PhD, acting deputy director of the Division of Monoclonal Antibodies at FDA, at a session on specification setting at the AAPS National Biotechnology Conference that was held June 19-21 in Boston.
Protein aggregation is a term that can include many types of aggregation, from rapidly reversible aggregation caused by non-covalent bonds to irreversible aggregation in the form of covalent oligomers.
On August 12, 2003, Johnson & Johnson began recalling certain batches of its anemia drug, Eprex (epoetin alfa, sold as Procrit in the US), in most countries outside of the United States.