



After reading Brian O'Connell's column in the February 2006 issue of BioPharm International, entitled, "Will Venture Capital Firms Turn Their Backs on Stem-Cell Research?" one would like to comment. The press is too fascinated by Dr. Hwang's scientific misconduct, which was discovered, and sanctions imposed.
Regulatory/GMP Compliance
Latest News

Advertisement
Advertisement
The Food and Drug Administration recently unveiled its long-awaited Critical Path Opportunities List, which maps out a number of "scientific projects" for improving the testing and production of biotech therapies. In its March report, FDA recognizes that problems in the characterization, testing, and quality management of medical products can delay clinical trials and even completely block drug development.

RFID is currently the most advanced tracking technology, but manufacturers need to pursue a multilayered approach . . .

In recent years, FDA has made positive moves to foster innovation by streamling regulation. But the agency's recent proposal to exempt manufacturing for phase 1 clinical trials from GMP requirements seems ill advised.

In the pharmaceutical industry, ultrafiltration (UF) membranes are used extensively in the downstream purification of recombinant proteins or monoclonal antibodies. However, the fouling of membranes after a unit operation?especially when recombinant proteins or monoclonal antibodies are highly concentrated?is a common problem. Typically, normalized water permeability (NWP) of a membrane can be reduced to about 20 percent of its original permeability at the end of an ultrafiltration-diafiltration (UF-DF) operation.

On January 17, 2006, the FDA released new regulations, effective June 1, 2006, which affect the production of most investigational drug and biologic products intended for phase 1 clinical trials. These regulations are much broader in scope than the Exploratory IND guidance released on the same day, and which apply only to low-risk, CDER-regulated clinical studies.

As part of its campaign to facilitate research on drugs and medical products, the Food and Drug Administration (FDA) recently issued new policies to encourage sponsors to conduct more informative and less costly early clinical trials. A new guidance on exploratory investigational drug applications (INDs) explains how scientists in industry and academia may test very small doses of a candidate compound to detect any pharmacologic effect before investing in more extensive in vitro and animal studies required for conventional phase 1 trials. The goal is to quickly identify products that show some promise of efficacy, and to halt research on those that fail to hit preliminary targets.

Signal-to-noise ratios are useful in robust engineering to design products and processes that consistently deliver on target.
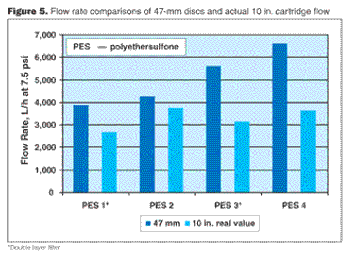
br> Sterilizing grade, 0.2-µm rated membrane filters are used in many biopharmaceutical processes to ensure the absence of particles and microorganisms from the filtered fluid (1, 2, 3). These filters must meet particular performance criteria in specifically defined applications. For this reason, during filter design, one performance criterion often is enhanced at the expense of another. Consequently, critical process and flow parameters must be defined appropriately to identify the optimal flow membrane filter for a specific application. This paper describes such an evaluation schematic and tests, as well as some common misconceptions.

Lyophilized, or freeze-dried, materials are challenging samples for quality assurance and quality control (QA/QC) measurement because of the inability to open the container without corrupting the product. Near-infrared analysis presents itself as the QC method of choice for lyophilized materials due to its ability to penetrate glass or plastic containers to analyze the sample in a non-destructive manner. This study demonstrates the performance of a Fourier transform near-infrared (FT-NIR) spectrometer used in analyzing lyophilized samples of thrombin, a topical coagulant commonly used in the medical and dental fields. Key stability parameters for lyophilized thrombin include moisture and potency, which can be predicted simultaneously from a single spectrum using multivariate analysis.
Development guidelines for MAbs serve as a blueprint for their manufacture, safety, and efficacy testing.

The Food and Drug Administration's Prescription Drug User Fee program (PDUFA) has to be reauthorized by Oct. 1, 2007, and all the interested parties are fine-tuning their wish lists for "improvements." Although some consumer advocates and their Congres-sional allies blast user fees for extending industry control over the drug approval process, FDA officials, pharma companies, and patient disease groups applaud the program's success in ending "drug lag" and speeding new drugs and biotech therapies to market.

Before designing cleaning procedures, it's vital to know all physical and chemical characteristics of the product ingredients.

Human infections with avian flu strain H5N1 are occurring in a number of southeast Asian countries that have experienced large outbreaks of avian influenza. How great a risk to the human population is posed by this virus, and what steps can be taken to minimize its impact? Preventive vaccines have great potential to avert the spread of avian flu and other infectious diseases. What are the factors affecting the creation of new vaccines, and how can they be optimized to promote public health?

Cleaning validation is a critical consideration in the pharmaceutical industry. Inadequate cleaning can result in contamination of drug products with bacteria, endotoxins, active pharmaceuticals from previous batch runs, and cleaning solution residues. Such contaminants must be reduced to safe levels, both for regulatory approval and to ensure patient safety.

A 2000 cyber crime study revealed that 71 percent of security breaches were caused by people who worked within the company.

For those conducting clinical trials, strict adherence to state regulatory requirements makes sound business sense.

Validation of a cleaning process demonstrates that it can reliably and effectively remove residue to an acceptable level.

Several methods are available to assess RNA integrity and purity, which may affect downstream applications.

Many industry professionals know that analytical testing for biopharmaceuticals for all raw materials, production in-process stages, and final containers must be validated, and they generally understand how this can be achieved. Many of us even understand the basic concepts of laboratory compliance and production process quality. However, how exactly are analytical test method performance and process robustness related and how do they depend on each other? Furthermore, how do we monitor and maintain the accuracy and reliability of analytical methods long after validation completion to ensure the suitability of these methods for measuring process quality?

...each day a product is delayed is estimated to cost upwards of $1 million to $100 million.

...biopharmaceutical companies still face rampant piracy and counterfeiting of patented products.

Current best practices of containment reduce the risks associated with biotech development.

The manufacturer should propose stability-indicating methodologies that provide assurance that changes in the identity, purity, and potency of the product will be detected.
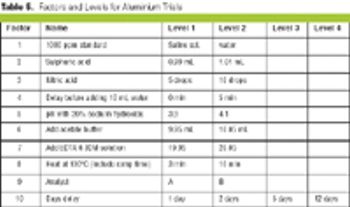
Saturated fractional factorial plans minimize the number of trials by one-half or better, which saves time and money.
Advertisement
Advertisement