


FDA has an official new leader, following a Senate vote to confirm Stephen Hahn as the agency’s next commissioner.
Quality/GMPs
Latest News
Advertisement
Digitalization of QbD Risk Assessments
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

Subjectivity in Quality Risk Management
The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Phase-appropriate Compliance for Cell and Gene Therapies
Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
Advertisement
The biologic is a novel bone builder that has a dual effect of increasing bone formation and reducing bone loss.
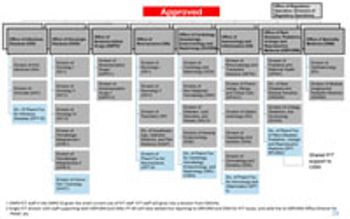
FDA’s Office of New Drugs restricting aims to improve scientific exchange and information sharing among review professionals.
A NASEM report stresses the importance of information sharing by biopharma companies and cooperation among regulatory authorities.
Accelerated approval pathways and growing demand for cell and gene therapies are putting pressure on providers of cellular starting materials, and they must ensure a steady supply.
Investigating deviations or failures of combination products needs to accommodate both the drug and device components, says Susan J. Schniepp, executive vice-president of post-approval pharma and distinguished
Problems in assuring reliable drug quality and supply dampens progress in bringing lifesaving therapies to market.
The agency sent a warning letter to Torrent Pharma after an inspection found violations of current good manufacturing practices that included a failure to thoroughly investigate batch failures.
The approval of Givlaari (givosiran) is the first for treatment of acute hepatic porphyria, which results in the buildup of toxic porphyrin molecules during the production of heme.
The agency’s approval of Abrilada (adalimumab-afzb), a biosimilar to Humira, brings the total of approved biosimilars to 25.
Roche’s ADC, Kadcyla, has been recommended for approval in the EU for for the adjuvant treatment of people with HER2-positive early breast cancer with residual invasive disease after neoadjuvant treatment.
Developers of biosimilars have become dismayed with difficulties in gaining acceptance and reimbursement from the US healthcare system.
In batch production, efficient exception management means reducing the time required to identify, review, and resolve process exceptions. Incorporating review by exception functionality within manufacturing execution system (MES) software can streamline biopharmaceutical product release.
In a statement, Janet Woodcock MD, director of FDA’s Center for Drug Evaluation and Research, highlighted the agency’s efforts to enhance the efficiency of postmarket drug safety surveillance.

Applying lessons of raw materials’ characterization and supply-chain control from the semiconductor industry allow more rigorous control of the biopharmaceutical manufacturing process.
The report focuses on information from stakeholders, published research, and economic analysis of market conditions from an analysis of drug shortage data and development of recommendations by the inter-agency Drug Shortage Task Force led by FDA.
Showing gains of 54% compared with 2018, results from a 2019 study showed that nearly three-quarters of pharma barcodes meet Drug Supply Chain Security Act requirements.
FDA readies more efficient oversight processes while advancing collaboration with Europe.

It is good industry practice to clarify the precise remit for each of the reviewers of a controlled document, says Siegfried Schmitt, PhD, vice-president, technical, Parexel Consulting.

Manufacturing differences between traditional mAb therapies and newer biotherapeutics dictate whether processes should be scaled up, scaled out, or use an alternate approach for commercial production.
The use of scale-down models allows for the theoretical optimization of processes and for troubleshooting problems during the developmental stage.
FDA report says drug shortages are underappreciated and patients pay a price.

Developing an effective bioassay is crucial for determining the potency of a drug substance or finished drug product. This article gives an overview of how to avoid most problems associated with correct bioassay development.
A recent report released by an FDA task force highlights the financial, manufacturing, and policy issues underlying the drug shortages of important prescription medicines in the United States.
The agency’s implementation of the SUPPORT Act has included new guidance documents and actions to restrict illicit drugs entering the US.
Advertisement
Advertisement