
Lonza revealed it received a warning letter from FDA for its cell therapy manufacturing plant.
Quality/GMPs
Latest News
Advertisement
Digitalization of QbD Risk Assessments
The digital transformation of quality-by-design assessment workflows can improve efficiency, reduce human errors, and facilitate integration within a much broader digital ecosystem.

Subjectivity in Quality Risk Management
The authors discuss subjectivity in the ICH Q9 (R1) guidance document.

Phase-appropriate Compliance for Cell and Gene Therapies
Understanding how to apply phase-appropriate GMPs is crucial for achieving successful regulatory approval.
Advertisement
The company is recalling one lot of 25% Dextrose injection after particulate matter was found within an internal sample syringe.
Sandoz, a Novartis division, announced that the Committee for Medicinal Products for Human Use (CHMP) has adopted positive opinions recommending the approval of its biosimilars rituximab and etanercept in Europe, for the same indications as the respective reference medicines.
The agency approved Renflexis, a biosimilar to Janssen’s blockbuster rheumatoid arthritis treatment.
The number of deficiencies found in foreign and UK-based facilities increased in 2016.
At CPhI North America 2017, the US Pharmacopeial Convention will be discussing its upcoming revision and modernization of the standards for elemental and organic impurities.
Pharmaceutical Technology spoke with CPhI North America presenter Jonathan Helfgott to discuss navigating GDUFA and helpful tips for submitting successful ANDAs.
The agency issued its recommendation for the influenza virus strains European vaccine manufacturers should include for 2017.
The new guide offers guidance on how to ensure data and records are complete, consistent, secure, accurate, and available throughout their lifecycle.
The agency cited the company’s India facility for batch failures and data integrity problems.
The agency released guidance on single assessments of PSURs to improve safety and benefit-risk assessment of medicines.
The role of patient advocates in shaping regulations and policy has put attention on financial and operational links between drug companies and independent health organizations.
FDA is in the center of the debate over developing and pricing new cancer therapies.
A new study in NEJM compares the regulatory review processes of FDA and EMA.
Scott Gottlieb answers Senators' questions at his confirmation hearing before the Senate Health, Education, Labor and Pensions Committee.
EMA has developed a framework and action plan to foster relationships with the academic community.
The company announced that Meridian Medical Technologies is extending a recall of EpiPen and EpiPen Junior to the United States.
The author discusses the impact of prefilled syringe product contact materials and the filling and stoppering process on protein aggregates.

As regulators strive for balance in cGMPs for cell, gene, and tissue therapies, risk-management principles must guide decisions involving process media and additives.
Susan Schniepp, distinguished fellow at Regulatory Compliance Associates, discusses the value of internal audits and how the information gained can be applied.
Industry fears limited benefits as FDA readies voluntary data tracking program.

The complex nature of biologics adds additional CQAs that must be determined to ensure the safe development of biologics
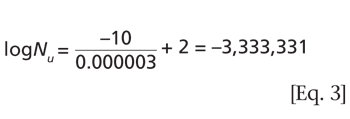
Understanding the purpose of the biological indicator can guide the development of an effective sterilization process.
The mAb is the first approved treatment that targets the progressive form of the disease.
The agency sent a warning letter to Opto-Pharm Pte Ltd. after deficiencies in sterile manufacturing procedures were found at the company’s Singapore facility.
Advertisement
Advertisement