




Departure from dilutional similarity can be interpreted as evidence that the groups of organisms are not comparable or the preparations do not contain the same active compounds.

Departure from dilutional similarity can be interpreted as evidence that the groups of organisms are not comparable or the preparations do not contain the same active compounds.

When data are not normal, a more efficient approach to monitor and control the performance of this assay requires transforming the data to a normal distribution. One of the most useful transformations was invented by Taguchi.

The manufacturer should propose stability-indicating methodologies that provide assurance that changes in the identity, purity, and potency of the product will be detected.
Synthetic drugs can be well characterized by established analytical methods. Biologics on the other hand are complex, high-molecular-weight products, and analytical methods have limited abilities to completely characterize them and their impurity profiles. Regulation of biologics includes not only final product characterization but also characterization and controls on raw materials and the manufacturing process.

Many types of equipment in both manufacturing and laboratory areas are critical to a properly functioning pharmaceutical process. The validation of laboratory equipment is not as clearly defined as the validation of equipment used directly in the production of pharmaceutical products, which requires thorough validation in almost all situations.

RARM procedures don't exist in a vacuum. For people to perform effective and useful RARMs, the process needs to be integrated with other GMP quality system elements and be proceduralized.

The purpose of design validation is to demonstrate that a product performs as intended. The usual route to this goal is showing that every item on the specification has been achieved, but it is not an easy path. The specification itself can create difficulty if it includes statements like "as long as possible" or the real horror "to be decided." Verification tests can reveal so many problems that the design must change to such an extent that earlier tests are no longer relevant. And there is also the practical difficulty of obtaining sufficient samples to test when the manufacturing engineers have not completed their standard operating procedures, the product design is not fixed yet, the component suppliers are late, and the marketing department has taken all the samples to show to prospective customers.

In order to institute a GxP mindset across the organization, support and respect for quality systems should come from the top down.
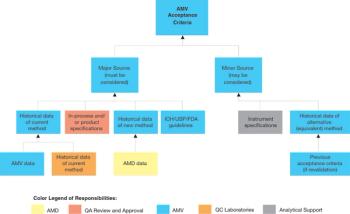
When replacing an existing method, it may be necessary to compensate for a significant difference between the current and new method by adjusting the specifications.

The relationship between "valid" or "suitable and validated" is often overlooked, but there is a high price when "validated" test systems are simply inappropriate.
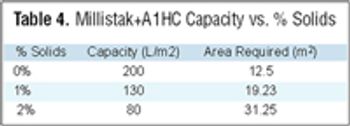
Filtration is one of the most commonly used unit operations in biopharmaceutical manufacturing. Available formats include direct or normal flow filtration (NFF) and cross or tangential flow filtration (TFF). These methods are used for sterilization and virus filtration, depth filtration or ultrafiltration, and diafiltration applications. Some common objectives include:

Networks are part of the compliance picture. Recent FDA warning letters show the agency considers network monitoring and qualification a necessary part of maintaining the security and integrity of electronic records.
The FDA?s risk-based approach to pharmaceutical cGMPs applies to 21 CFR Part 11 enforcement as well. Understanding different methodologies for assessing and managing risk will help you develop and begin to implement a compliance plan.
Protecting the integrity of data is a challenge of 21 CFR Part 11 compliance. Integrity requires records to be complete, intact, and maintained in their original context ? associated with the procedures which were used to create the data.
Bringing different laboratory instruments into compliance takes planning. The key strengths and weaknesses of different levels of control and feedback for analytical instruments and data transfer systems are highlighted in this article.

This article focuses on the front end of qualifying a new raw material from a given supplier. Once qualified, this status must be maintained by periodic review and requalification.

Biotech companies are among the most intensive instrumentation users of any FDA-regulated business. Because of the complex nature of these companies' research and manufacturing, they typically have a larger number of instruments per employee.