





Now equipment makers are designing new systems that can be deployed rapidly, are less costly to build, and require less water and power to operate.
Now equipment makers are designing new systems that can be deployed rapidly, are less costly to build, and require less water and power to operate.
FDA's approach involves adopting efficient strategies for targeting inspections to more high-risk operations likely to have the greatest impact on public health.
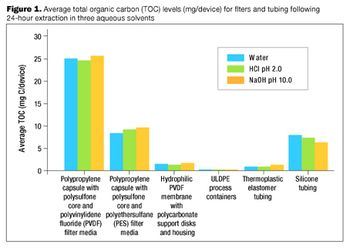
The many benefits of disposable technologies, such as significant savings in time, labor and capital, as well as ease of scalability and flexibility, have led to the growing trend of adopting disposable technologies in bioprocess manufacturing processes.

The first part of this article, published in the September 2006 issue, discussed general strategies for validation extensions to other test method components, laboratories and even different test methods.1This second part provides practical tips on how to maintain test method suitability long after the formal completion of analytical method validation (AMV) studies.

Before designing cleaning procedures, it's vital to know all physical and chemical characteristics of the product ingredients.

Cleaning validation is a critical consideration in the pharmaceutical industry. Inadequate cleaning can result in contamination of drug products with bacteria, endotoxins, active pharmaceuticals from previous batch runs, and cleaning solution residues. Such contaminants must be reduced to safe levels, both for regulatory approval and to ensure patient safety.

Many industry professionals know that analytical testing for biopharmaceuticals for all raw materials, production in-process stages, and final containers must be validated, and they generally understand how this can be achieved. Many of us even understand the basic concepts of laboratory compliance and production process quality. However, how exactly are analytical test method performance and process robustness related and how do they depend on each other? Furthermore, how do we monitor and maintain the accuracy and reliability of analytical methods long after validation completion to ensure the suitability of these methods for measuring process quality?
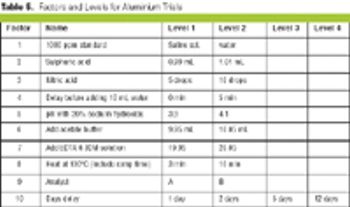
Saturated fractional factorial plans minimize the number of trials by one-half or better, which saves time and money.
Synthetic drugs can be well characterized by established analytical methods. Biologics on the other hand are complex, high-molecular-weight products, and analytical methods have limited abilities to completely characterize them and their impurity profiles. Regulation of biologics includes not only final product characterization but also characterization and controls on raw materials and the manufacturing process.

Many types of equipment in both manufacturing and laboratory areas are critical to a properly functioning pharmaceutical process. The validation of laboratory equipment is not as clearly defined as the validation of equipment used directly in the production of pharmaceutical products, which requires thorough validation in almost all situations.

The purpose of design validation is to demonstrate that a product performs as intended. The usual route to this goal is showing that every item on the specification has been achieved, but it is not an easy path. The specification itself can create difficulty if it includes statements like "as long as possible" or the real horror "to be decided." Verification tests can reveal so many problems that the design must change to such an extent that earlier tests are no longer relevant. And there is also the practical difficulty of obtaining sufficient samples to test when the manufacturing engineers have not completed their standard operating procedures, the product design is not fixed yet, the component suppliers are late, and the marketing department has taken all the samples to show to prospective customers.
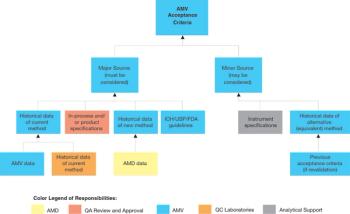
When replacing an existing method, it may be necessary to compensate for a significant difference between the current and new method by adjusting the specifications.

The relationship between "valid" or "suitable and validated" is often overlooked, but there is a high price when "validated" test systems are simply inappropriate.
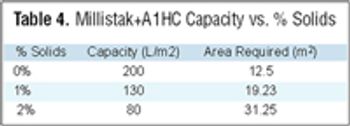
Filtration is one of the most commonly used unit operations in biopharmaceutical manufacturing. Available formats include direct or normal flow filtration (NFF) and cross or tangential flow filtration (TFF). These methods are used for sterilization and virus filtration, depth filtration or ultrafiltration, and diafiltration applications. Some common objectives include:

Networks are part of the compliance picture. Recent FDA warning letters show the agency considers network monitoring and qualification a necessary part of maintaining the security and integrity of electronic records.

Biotech companies are among the most intensive instrumentation users of any FDA-regulated business. Because of the complex nature of these companies' research and manufacturing, they typically have a larger number of instruments per employee.