


The opening presentation gives the company a chance to put their best foot forward, according to Siegfried Schmitt, principal at PAREXEL.
Process Validation
Latest News
Advertisement
Advertisement

Can bioprocessing runs be consistently replicated in an inherently variable production environment?
Lonza revealed it received a warning letter from FDA for its cell therapy manufacturing plant.
The new guidelines will address bioanalytical method validation and biopharmaceutics classification system-based biowaivers.
The author provides a review of the concepts of design and qualification that apply to single-use systems.
The agency publishes draft guidance on assay development and validation for immunogenicity testing.
While stakeholders generally welcome improvements to quality initiatives, they are concerned with how the new requirements will be implemented for more complicated supply-chain models.

Can alternative approaches to the permitted/acceptable daily exposure be justified?
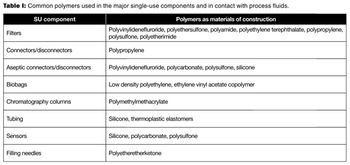
The author discusses the current best practices in technical qualification of single-use systems.
Regulatory officials and industry scientists participated in a CMC Strategy Forum sponsored by CASSS in July 2015.
This article discusses cleaning validation of equipment dedicated to the production of a single API.
The guidance provides recommendations for submitting analytical procedures and method validation data to FDA.
Single-use and modular technologies plus continuous manufacturing are increasingly important to biopharma scale-up and tech transfer.
Guidance provides labeling standards and an implementation guide for electronic submission of lot distribution reports.
Evaluating the assembly design process, manufacture, and use helps mitigate risk.

The author presents opportunities and challenges in implementing the product lifecycle approach.
GS1 publishes a healthcare industry guideline describing how to implement GS1 standards to support requirements of the 2013 US Drug Supply Chain Security Act.
The BioPhorum Operations Group has published a resource to help the biopharmaceutical industry deliver a consistent approach to continued process verification.
FDA cites cGMP violations for API manufacturing at a facility in Tianjin, China.
FDA inspection found cGMP violations in the Bangalore, India API manufacturing facility.

Experts give insight on method transfer, QbD, and regulations for analytical method development and validation for biopharmaceuticals.
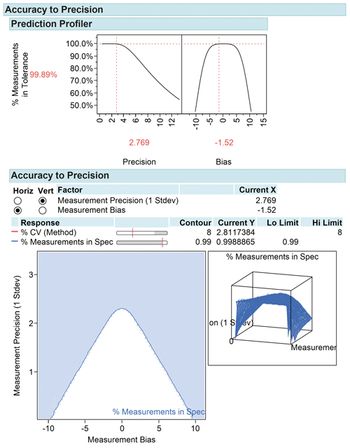
Design of experiment is a powerful development tool for method characterization and method validation.
Thirteen companies are accepted for participation in the supply chain program.

The European Union is strengthening its pioneering role in the regulation of biosimilars by further developing the basic rules for determining the levels of compatibility for this group of drugs. There are, however, some key issues that are not easy to resolve, as evident in a recent workshop on biosimilars organized by the European Medicines Agency (EMA).

The rising cost of drug development and the decreasing proportion of drug-naive population in the US and European markets are driving international pharmaceutical companies to consider emerging markets as a location to conduct their clinical trials. Asia stands out among the emerging markets given its double-digit growth rates.
Advertisement
Advertisement