FDA issues a warning letter to Megafine Pharma Limited for data integrity violations.
Regulatory/GMP Compliance
Latest News
Advertisement
Advertisement
The agency provides guidelines on generating histopathology data in nonclinical biomarker qualification studies.
Biomedical innovation legislation may stall in 2016.
Progress is being made, but work remains to be done. Brian Daleiden, vice president of marketing at TraceLink, discusses the state of the industry, as companies attempt to meet complex, and changing, global requirements.
Siegfried Schmitt, Principal Consultant, PAREXEL International, discusses how to report quality metrics to FDA.
The guidance discusses the implementation of CMC postapproval changes through a comparability protocol.
FDA explains the rewards that may be associated with switching from batch to continuous manufacturing.
Jack Lew, Obama’s secretary of the treasury, announced on April 4, 2016, that the US Department of the Treasury and the Internal Revenue Service (IRS) is issuing temporary and proposed regulations to limit the “benefits of and limit the number of corporate tax inversions.” The government bodies also plan to address earnings stripping in these inversions, so it will examine past inversion deals that have already been completed.
The draft guidance outlines ways applicants can test for abuse deterrence in solid oral opioid drugs.
The agency now allows production of water for injection by non-distillation technologies.

Data integrity is a widespread, global problem that must be addressed.
The agency published guidance on immunogenicity-related considerations for low molecular weight heparin.
The directorate is looking for experts to join the European Pharmacopoeia network.
The agency is continuing CDER’s Regulatory Project Management Site Tours and Regulatory Interaction Program.
The European Directorate for the Quality of Medicines & Healthcare announces the publication of a chemometric methods chapter in the European Pharmacopoeia.
FDA’s Center for Drug Evaluation and Research publishes a list of upcoming new and revised guidance documents for 2016.
FDA issued a warning letter to Chan Yat Hing Medicine Factory for CGMP violations.
The European Pharmacopoeia rewrites in its general chapter on Raman spectroscopy.
Three associated centers in California, New York, and Florida are cited for not following proper sterilization and validation procedures and for not having the appropriate licenses to manufacture a biological drug product.

Traceability and transparency will remain elusive if manufacturers continue to approach serialization projects on a case-by-case basis.
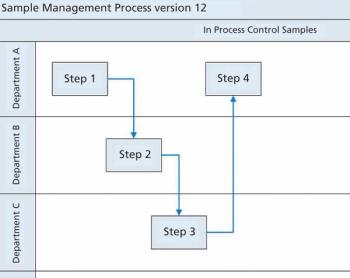
Siegfried Schmitt, principal consultant, PAREXEL, discusses how to streamline the document management process during market expansion.
The United States Pharmacopeial Convention announced the recipients of the 2015–2016 Global Fellowship Awards.
Finalizing GMP requirements and quality standards for the development, manufacture, and clinical testing of ATMPs in the EU is proving to be a complex task.
FDA seeks feedback on possible analytical standards and approaches to optimize regulation of next-generation sequencing (NGS)-based in vitro diagnostic tests.
Diversifying the Global Heparin Supply Chain: Reintroduction of Bovine Heparin in the United States?
The global supply chain for bovine and porcine heparin and regulatory considerations are examined.
Advertisement
Advertisement