



Fruzaqla was previously approved for use in patients with metastatic colorectal cancer in the US in November 2023.
Fruzaqla was previously approved for use in patients with metastatic colorectal cancer in the US in November 2023.

FDA has granted expanded approval to Sarepta Therapeutics for Elevidys to treat DMD in non-ambulatory patients, in addition to ambulatory patients.
Immunology company argenx gets its third FDA-approved indication for VYVGART Hytrulo for treating chronic inflammatory demyelinating polyneuropathy.

Imfinzi (durvalumab) combined with chemotherapy decreased the risk of disease progression or death by 58% in a 700-patient trial.
FDA’s priority review status was granted based on positive results from a Phase III study evaluating Sarclisa in combination with VRd in treating transplant-ineligible newly diagnosed multiple myeloma.
Bkemv (eculizumab-aeeb) is the first interchangeable biosimilar to Soliris (eculizumab) to treat paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome.
Beqvez (fidanacogene elaparvovec-dzkt), a one-time gene therapy, helps adults with hemophilia B produce factor IX themselves instead of receiving regular intravenous infusions.

GSK’s supplemental Biologics License Application for its PD-1-blocking antibody therapy has been accepted for review by FDA.
J&J’s nipocalimab is in development for reducing the risk of FNAIT in alloimmunized pregnant adults, a rare disease that may risk the life of the fetus or newborn.
AstraZeneca notes that Ultomiris is the first and only long-acting C5 complement inhibitor that offers NMOSD patients the potential to live without relapsing.
AbbVie’s acquisition of Landos includes a lead asset that boosts its portfolio in autoimmune and inflammatory diseases, while the ADC, ELAHERE, gets full FDA approval.
Orchard Therapeutics’ Lenmeldy (atidarsagene autotemcel) marks the first gene therapy approved in the US for treating children with metachromatic leukodystrophy.
EMA has validated two MAAs submitted by AstraZeneca and Daiichi Sankyo for datopotamab deruxtecan in two types of cancer.
The agency has recommended granting marketing authorization to ALS treatment, Qalsody (tofersen), for adults who have a mutation in the superoxide dismutase 1 (SOD1) gene.

Daiichi Sankyo is investing approximately €1 billiion (US$1.08 billion) to expand its Pfaffenhofen an der Ilm, Germany, site for ADC development and production.
FDA has approved Iovance Biotherapeutics’ Amtagvi (lifileucel) for treating patients with unresectable or metastatic melanoma.
The three non-commercial developers of ATMPs will benefit from enhanced support from the agency.
NICE has recommended the use of AbbVie's Tepkinly as a treatment option for eligible adults with diffuse large B-cell lymphoma.

In the ATMP space, CGTs are hitting their stride with unprecedented approvals in the past year alone.
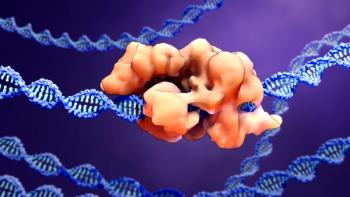
The milestone approval of a gene-edited therapeutic paves the way for gene-editing technologies.
The final guidance provides specific recommendations for CMC, pharmacology, toxicology, and clinical study design for CAR-T cell products.
The final guidance provides recommendations for developing gene therapy products incorporating genome editing of human somatic cells.
Takeda has received FDA approval for HYQVIA, a subcutaneous immunoglobulin for the maintenance treatment of chronic inflammatory demyelinating polyneuropathy.
The agency recommended 77 drugs for marketing authorization in 2023, including 39 new APIs.
FDA’s Center for Drug Evaluation and Research released its report on new drugs approved in 2023.