The agency sent warning letters to three more companies for selling unapproved products claiming to treat COVID-19.
Regulatory/GMP Compliance
Latest News
Advertisement
Advertisement
EMA's CHMP has recommended the use of remdesivir, an investigational antiviral medicine, in compassionate use programs across the European Union.
Addressing data integrity, quality culture, aging facilities, investigations/corrective actions and preventive actions, and risk management is key when conducting audits, says Susan J. Schniepp, executive vice-president of post-approval pharma and distinguished fellow, Regulatory Compliance Associates.
With new tests showing NDMA levels increase under normal storage conditions, FDA calls for removal of ranitidine products from the market.
FDA is encouraging alternative insulins and challenging anticompetitive practices.

Risk assessments, audits, and good communication between sponsor and supplier are key elements of supplier oversight.
The bill includes multiple less-noticed provisions to bolster healthcare programs and to advance the development of new treatments and preventives to combat the virus.
Noting traditional clinical trials for COVID-19 convalescent plasma will take time, FDA is allowing physicians to submit requests for single-patient emergency INDs.
In light of the current COVID-19 pandemic, the agencies co-chaired the first global regulators meeting to facilitate development of vaccines against SARS-CoV-2, which causes COVID-19.
FDA officials are rolling out guidance and support for researchers striving to assess potential treatments for COVID-19 while the agency tries to object to premature optimism and regain public credibility.
FDA has been working closely with other government agencies and academic centers that are investigating the use of the drug chloroquine to determine whether it can be used to treat patients with mild-to-moderate COVID-19.
FDA is offering advice and added flexibility to help sponsors adjust ongoing and planned clinical research programs during the COVID-19 outbreak.
FDA postpones routine domestic facility inspections due to the COVID-19 pandemic.

This article details the more operational aspects of monograph submissions, answering the question of how to participate.
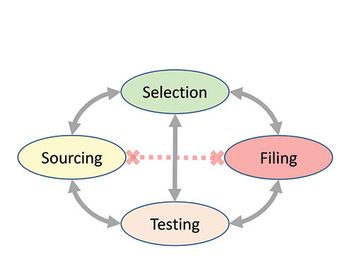
The authors present a case study with raw materials and excipients, where a consistent, cross-functional approach is needed to ensure the appropriate selection, sourcing, testing, and filing of the materials used to manufacture bio/pharmaceutical products in a global environment, ensuring compliance with applicable compendial and regulatory requirements.

This series is intended to address the challenges for the industry to comply with pharmacopoeial requirements. This article returns to this important topic with a case study at the intersection of monograph development and compliance.

This article summarizes all the considerations that go into a company’s compendial affairs program and to look ahead at topics that will likely result in further evolution in the pharmacopoeias around the world. This look into what is on the horizon is important to help companies prepare for the inevitable changes and ensure the continued supply of quality medicines to patients globally.

Find links to pertinent regulatory and standard setting resources, guidance documents, and guidelines.

Connect with pharmaceutical and healthcare regulatory authorities around the world via this directory.

With some FDA inspections on hold, will the US drug supply maintain its quality standards?

Monographs are developed based on the submission of information and materials from a company having regulatory approval for the product, and this submission feeds into the pharmacopoeia revision process.

This final article in the series has two purposes: to summarize all the considerations that go into a company’s compendial affairs program and to look ahead at topics that will likely result in further evolution in the pharmacopoeias around the world.

This article returns to the topic of complying with pharmacopoeial requirements with a case study at the intersection of monograph development and compliance.

In January 2020, the agency finalized six clinical development and manufacturing guidance documents and drafted new guidance on what would qualify new gene therapies as orphan drugs.
FDA issued a notice to drug compounders regarding the transition of licensure of biologics to the Public Health Service Act.
Advertisement
Advertisement