

Formulation
Latest News
Advertisement
Latest Videos





Advertisement
More News

With $137 million in Series B funding led by Novartis and other firms, Sidewinder Therapeutics will support development of bispecific ADCs designed to improve tumor targeting, enhance drug delivery, and reduce off-target toxicity in cancer.

Experts from Acuitas, Mana.bio, NanoVation, and ReCode share their input on targeted LNP engineering, which is accelerating pipeline expansion of nucleic acid therapies for chronic and rare diseases beyond the liver.
FDA’s breakthrough therapy designation for Johnson & Johnson’s co-formulated bispecific antibody therapy validates dual EGFR/MET targeting in HPV-negative head and neck cancer.

PharmaResearch’s DOT-based nanoparticle platform enters US clinical testing, highlighting delivery innovation aimed at improving tolerability in solid tumor therapies.

NeoVac first-in-human data suggest that optimized lipid nanoparticles may improve mRNA tolerability, enabling repeat dosing and broader therapeutic use.

Study data show high-dose nusinersen can improve function and slow neurodegeneration, informing future SMA dosing strategies and lifecycle management.

The approval of aflibercept for visual impairment from macular edema following retinal vein occlusion is based on clinical data that suggests higher-dose anti-VEGF therapy can preserve vision while extending dosing intervals.
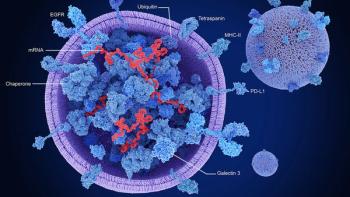
Engineered exosomes could reshape therapeutic development by redefining delivery, manufacturing models, and regulatory standards.

The new collaboration will aim to streamline formulation-to-manufacturing workflows and de-risk development.

The company demonstrates advances in glioma hydrogel, high-concentration biologics, and nanoencorafenib licensing, impacting drug delivery strategies.

Non-parenteral alternatives for biologics remain a clinical imperative and a formidable challenge.
This quiz measures your comprehension of one of our recent feature articles.
This quiz measures your comprehension of one of our recent feature articles.
A growing demand for liquid medicines is driven by patient groups' unique needs, improving compliance through flexible and palatable dosing options.

Formulation and analytics are combining to advance drug development synergistically, with evolving tools and related strategies shaping quality, scalability, and innovation.

The industry is diversifying pipelines from traditional small-molecule drugs to embrace complex and exciting new modalities.

The spray-dried formulations for Ethris’ mRNA vaccine candidates will be developed at Lonza’s Bend, Ore., Center of Excellence in accordance with GMP standards.
Exosomes, polymeric nanoparticles, and DNA nanostructures offer many potential advantages.

Takeda’s TAKHZYRO (lanadelumab) is now approved in Europe as a subcutaneous injection treatment for hereditary angioedema in patients 12 years old and above and in adults.
Lonza will develop spray-dried formulations for an intranasally delivered biologic using a reformulated biologic drug candidate for obesity in Iconovo’s pipeline.

Using Orexo’s powder-based drug delivery technology, the companies will develop mucosal vaccines in an inhaled formulation.
The companies will develop a platform that can enable rapid development of DPI products.

Lonza’s new tailored offering leverages the company’s bi-layer capsule manufacturing technology.
Under the collaboration, the companies will create and test circVec DNA–LNP formulations with an eye toward potential therapeutic applications.

Solving the challenge of better-stabilized temperature-sensitive biomolecules hinges on innovative formulation strategies.
Advertisement
Advertisement