

The European Medicines Agency's Annual Report highlights the agency's drug approvals, projects, and initiatives for 2013.
Regulatory Authority Actions
Latest News
Advertisement
Advertisement
EMA notifies EU healthcare professionals of the falsified cancer drug Herceptin.

New identifiers and tracking requirements aim to block illegitimate products.
Looking to improve patient access to new medicines, EMA creates a pilot project to explore an adaptive licensing approach with real medicines in development.
FDA clarifies recommendations for injectable drug products packaged in vials and ampules.
HHS plan makes progress in ensuring availability of safe vaccines.
Agencies extend successful pilot program to further harmonization of QbD topics.
New guidance from FDA asks for documentation of CMC postapproval manufacturing changes.
Accelerated testing and production create challenges in documenting product quality.

China's regulatory and compliance environment is set to change as the government declares a crackdown on bribery scandals.
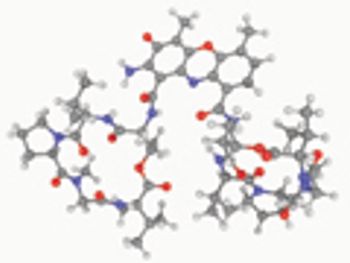
USP evaluates quality attributes for synthetic peptides.

Manufacturers are taking measures to comply with new package safety rules.
FDA revises interpretation of the five-year NCE exclusivity provisions for certain fixed-combination drug products.
FDA provides recommendations for submitting analytical procedures and methods validation data.
FDA and EMA set up regular meetings to harmonize drug safety actions.
Thirteen companies are accepted for participation in the supply chain program.
EMA releases an update on its flu vaccine guidance.
EMA and the European Commission revise Q&A document on implementation of marketing authorization guidelines.
FDA plans to issue a number of new guidances in 2014 that will address drug development and manufacturing practices.
NIH, 10 biopharmaceutical companies, and several nonprofit organizations form a partnership to develop new treatments earlier for Alzheimer's, type 2 diabetes, and autoimmune disorders.
High technology assessments are having an impact on biosimilars development in Europe.
EMA's guidance focuses on the use of pharmacogenomics to improve drug safety monitoring.
FDA adds Ranbaxy's Toansa, India facility to existing consent decree, prohibiting distribution of APIs from that location
FDA releases guidance on developing drugs for the treatment of community-acquired bacterial pneumonia.
FDA releases guidance on the qualification of drug development tools.
Advertisement
Advertisement