FDA sent a warning letter to GPT Pharmaceuticals Pvt. Ltd. after inspectors found CGMP violations that included equipment that was not properly maintained.
Regulatory Authority Actions
Latest News
Advertisement
Advertisement
Pressures on FDA will affect industry’s success in bringing new therapies to market.
FDA sent a warning letter to Dercher Enterprises, Inc., DBA Gordon Laboratories, for CGMP violations and adulterated drug products.
The guidance describes procedures for obtaining an additional National Drug Code for prescription drugs imported into the United States.
The European Medicines Agency and its European partners have launched a pilot program for cooperation in the inspection of facilities that manufacture sterile drug products.

Warning letters tell the tale of missteps by drug companies and offer a path to compliance for quality teams that monitor these enforcement actions.
Production and process controls, organization and personnel were the top problems found, while packaging and labeling citations increased in 2017 and 2018.
According to a consent decree of permanent injunction issued in September 2019, the health and wellness companies must recall and stop distributing products until the companies comply with the Federal Food, Drug, and Cosmetic Act and other requirements included in the consent decree.
The biologic is a novel bone builder that has a dual effect of increasing bone formation and reducing bone loss.
The app, CURE ID, is designed to allow the clinical community to report their experiences treating infectious diseases with novel uses of existing FDA-approved drugs.
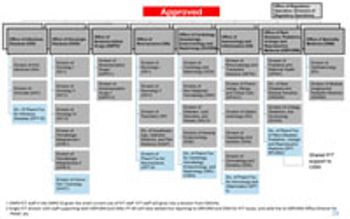
FDA’s Office of New Drugs restricting aims to improve scientific exchange and information sharing among review professionals.
A NASEM report stresses the importance of information sharing by biopharma companies and cooperation among regulatory authorities.
The agency sent a warning letter to Torrent Pharma after an inspection found violations of current good manufacturing practices that included a failure to thoroughly investigate batch failures.
The approval of Givlaari (givosiran) is the first for treatment of acute hepatic porphyria, which results in the buildup of toxic porphyrin molecules during the production of heme.
The agency’s approval of Abrilada (adalimumab-afzb), a biosimilar to Humira, brings the total of approved biosimilars to 25.
FDA issued a warning letter to the company for obtaining over-the-counter drugs made by foreign manufacturers that violated federal law.
Roche’s ADC, Kadcyla, has been recommended for approval in the EU for for the adjuvant treatment of people with HER2-positive early breast cancer with residual invasive disease after neoadjuvant treatment.
Oncologist Stephen Hahn has been nominated for the top post, following the appointment of Brett Giroir as acting commissioner.
In a statement, Janet Woodcock MD, director of FDA’s Center for Drug Evaluation and Research, highlighted the agency’s efforts to enhance the efficiency of postmarket drug safety surveillance.
The report focuses on information from stakeholders, published research, and economic analysis of market conditions from an analysis of drug shortage data and development of recommendations by the inter-agency Drug Shortage Task Force led by FDA.
Showing gains of 54% compared with 2018, results from a 2019 study showed that nearly three-quarters of pharma barcodes meet Drug Supply Chain Security Act requirements.
FDA report says drug shortages are underappreciated and patients pay a price.
A recent report released by an FDA task force highlights the financial, manufacturing, and policy issues underlying the drug shortages of important prescription medicines in the United States.
The agency’s implementation of the SUPPORT Act has included new guidance documents and actions to restrict illicit drugs entering the US.
The agency announced it has approved 1171 generic drugs in fiscal year 2019.
Advertisement
Advertisement