




Transcriptomics plays a role in influencing the production of recombinant therapeutics in microbial and mammalian hosts.
Transcriptomics plays a role in influencing the production of recombinant therapeutics in microbial and mammalian hosts.

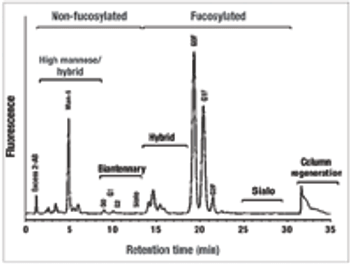
The authors review how media components modulate the quality of monoclonal antibody products

A novel coiled flow inversion reactor (CFIR) improves process productivity and performance.

The authors review the technologies that may help bioprocessing become a truly continuous operation and present case studies that could contribute to the integration of upstream and downstream platforms.
The authors review the status of expression of antibodies in microbial hosts and present the recent advances in the production of aglycosylated antibodies in bacteria.
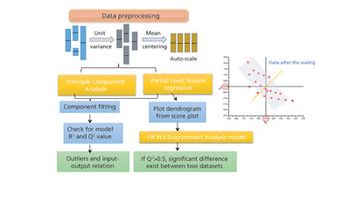
The authors review major developments in use of MVDA in bioprocessing applications.

Large-scale implementation of Protein A chromatography offers several challenges.

The authors take a look at the past and future impact of the Indian Pharmacopoeia Commission.
Establishing the CQAs of a mAb product by evaluating impact and uncertainty during risk assessment.
The authors discuss the evolution of the purification platform for manufacturing of mAb therapeutics.
The authors discuss the application of risk management in process lifecycle validation, manufacturing, and change control.

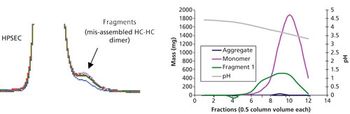
Aggregate formation is influenced by multiple aspects of the bioproduction process but can be mitigated by good process design and control.

The authors demonstrate how an integrated model is helping to achieve regulatory flexibility. This article is part of a special section on biopharmaceutical trends.
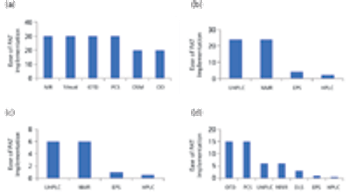
The authors review the various analytical methods that can enable use of PAT.

Challenges of vaccine development include regulatory, technical, and manufacturing hurdles in translating a vaccine candidate into a commercial product.
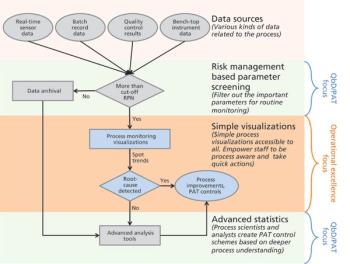
The authors focus on operational excellence in manufacturing of biotechnology therapeutic products in the QbD paradigm.
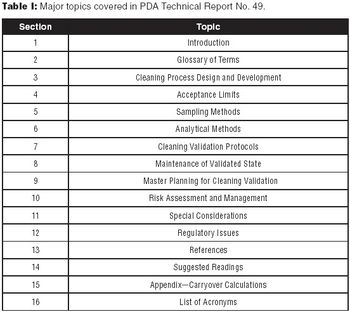
The authors encourage biotech manufacturers to consult PDA Technical Report No. 49 for a detailed perspective on current practices and issues in biotech cleaning validation.

An approach to reduce batch time, increase productivity, and decrease costs.

Evaluate and communicate risk to stakeholders.
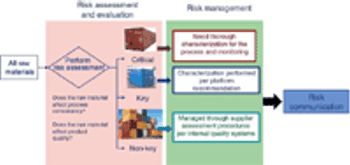
Adequate characterization of materials protects product quality.

Regulatory flexibility can make continuous improvement possible.
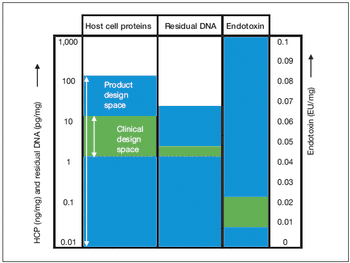
Key considerations for defining your overall control strategy.
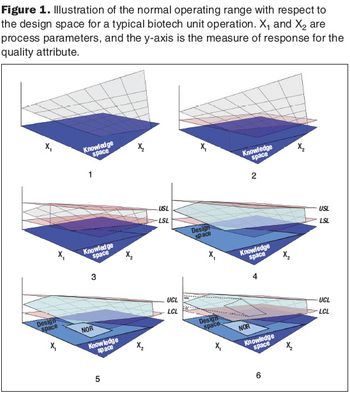
Second in a three-part series that discusses the complexities of QbD implementation in biotech development.
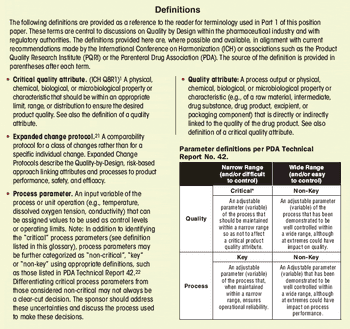
First in a three-part series that discusses the complexities of QbD implementation in biotech product development.
Select the best approach to determine critical quality attributes.