




Phenomenex opened a new manufacturing and development facility in CA for the company’s gas chromatography columns.
Biopharmaceutical Analysis: GC-MS
Latest News
Advertisement
Advertisement
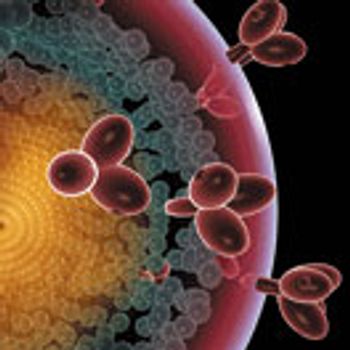
This article summarizes the approaches, challenges, and future perspectives for the characterization of N-glycans in biopharmaceutical products.

Extraction studies demonstrate approaches for evaluating single-use bio-pharmaceutical manufacturing materials.
The Chinese facility was cited for data integrity violations.

A step-wise process is used to characterize glycans and understand the functioning of a molecule for biosimilar development.
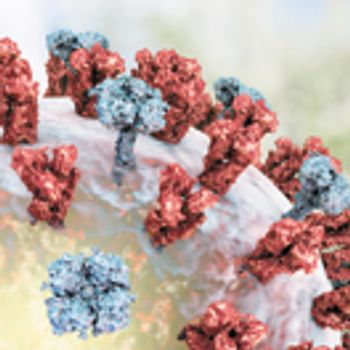
The authors present a review of the techniques commonly used for glycosylation analysis.

Industry experts spoke to BioPharm International about the key considerations in the development of a drug-delivery device for a biologic drug, the importance of human factors engineering, the advantages of prefilled syringes, and the challenges in the manufacture of these devices.

The author discusses the various ways in which a quality-by-design program can enhance the extractable and leachable assessment of a drug product.
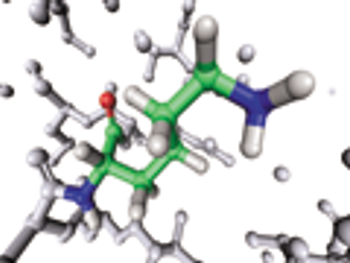
USP evaluates raw materials used in the chemical synthesis of peptides.
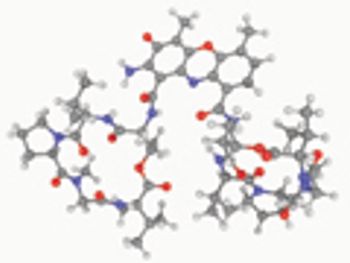
USP evaluates quality attributes for synthetic peptides.
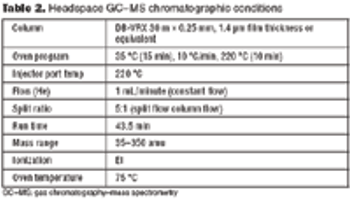
A systematic approach facilitates formulation component selection.
Advertisement
Advertisement