

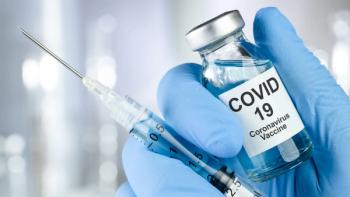
Low- and middle-income countries are at the greatest risk of receiving substandard and falsified COVID-19 vaccines.
Low- and middle-income countries are at the greatest risk of receiving substandard and falsified COVID-19 vaccines.
BlackBerry’s QNX real-time operating system may create cybersecurity vulnerabilities, according to FDA.
The agency is endorsing recommendations for global track-and-trace systems developed by the International Coalition of Medicines Regulatory Authorities.

The industry has made great strides in implementing serialization technologies, while technology developers are enabling more open access to real-time transportation data.
FDA, US Customs and Border Protection, and the US Immigration and Customs Enforcement, Homeland Security Investigations signed a Memorandum of Understanding to prevent illegal drug imports from entering the US through International Mail Facilities.
A strategic serialization partnership has been announced by advanco and Syntegon, aimed at tackling the global issue of counterfeit drugs.
TraceLink will participate in the agency’s Pilot Project Program under the Drug Supply Chain Security Act.
Supply chain players are eligible to participate in FDA’s pilot program to develop an electronic system for tracking drug products.
Efforts to establish an international system for identifying and tracing drugs through the global supply chain are moving forward.
The agency will lead an international team to create a Supply Chain Security Toolkit of resources to educate the pharmaceutical and healthcare industries on supply chain vulnerabilities.

As the November 2017 deadline nears, a surprising number of companies still don’t have a serialization plan in place. New programs aim to get them compliant in time.
The companies have developed a Level 4 traceability solution to manage pharmaceutical regulatory requirements.
Interactive Coding Equipment has produced a detailed guide to help pharmaceutical manufacturers make informed choices in the selection of suitable coding equipment for their applications.
TraceLink launched the EU-hosted early access program to reduce the time, cost, and risk of achieving regulatory compliance for global requirements.
A report from the European Union Intellectual Property Office shows that the EU loses approximately €10.2 billion a year due to counterfeit medicines.
Mumbai, the location for its new Indian subsidiary, will also be the site for the company's Uniquity Global Conference on October 6, 2016.
Progress is being made, but work remains to be done. Brian Daleiden, vice president of marketing at TraceLink, discusses the state of the industry, as companies attempt to meet complex, and changing, global requirements.
The agency prepares a plan to implement new packaging safety features.

Virtual pilot programs examine scenarios that may occur while implementing serialization requirements for the US Drug Supply Chain Security Act.
UPS joins the Global Health Supply Chain Technical Assistance program to help secure the drug supply chain.
FDA and industry support global framework and collaborations to secure the supply chain.
Pharmacy associates say more education regarding the transactional requirements of DSCSA is needed.
The agency takes action against websites that illegally sell unapproved medications.
The biopharma industry has had some successes but still needs to work on operational issues.
The agency is enlisting members of European pharmaceutical associations and industry representatives to participate in IDMP task force.