
The company has officially opened a local brand office in South Korean to support its existing business in the region.
Manufacturing, Liquid Drug Products
Latest News
Advertisement
Advertisement
The $72-million investment, part of a larger $850-million investment into its US operations, will allow the drugmaker to replace an outdated insulin vial-filling line and to upgrade technology at its Indianapolis manufacturing plant.
Stainless-steel particulates were found in one lot of 0.9% Sodium Chloride Injection, USP 1000 mL, prompting the recall to the hospital/user level.
Operated by BioOutsource, Sartorius’ subsidiary, the Glasgow, UK-based service center will offer physicochemical properties and structural attributes testing and allow clients to perform structural and functional analyses in parallel.

The unique structures of fusion proteins lead to expression, heterogeneity, and stability issues.
Hospira issued a voluntary worldwide recall due to confirmed subpotency and elevated impurity levels.
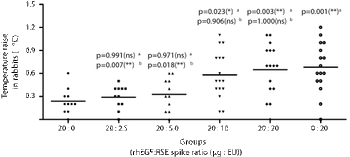
The correlation between limulus amebocyte lysate (LAL) assay and rabbit pyrogen test (RPT) targeting recombinant human epidermal growth factor (rhEGF) as active molecule was assessed.
A Q&A with SCHOTT Pharmaceutical Systems and West Pharmaceutical Services
Advertisement
Advertisement