



This article details the more operational aspects of monograph submissions, answering the question of how to participate.
Pharmacopoeial Compliance Series
Latest News
Advertisement
Advertisement
This case study is based on the experience of one of the authors but is applicable to all companies across the broader industry, illustrating the potentially surprising point that some compliance difficulties may be of the company’s own making.
This article explores another proactive advocacy approach that a bio/pharmaceutical company may take through participation in the development of new and revised monographs in the various pharmacopoeias.
This final article in the series has two purposes: to summarize all the considerations that go into a company’s compendial affairs program and to look ahead at topics that will likely result in further evolution in the pharmacopoeias around the world.
This article presents a case study at the intersection of monograph development and compliance.

The revision process and the resulting publication of proposed and official updates for pharmacopoeias around the world are described.

This article describes the revision process and the resulting publication of proposed and official updates for pharmacopoeias around the world.

An effective surveillance program for monitoring the activities of pharmacopoeias around the world requires processes, people, and tools from across a company.

An effective surveillance program for monitoring the activities of pharmacopoeias around the world requires processes, people, and tools from across a company.
An understanding of global and national pharmacopoeias is crucial to understanding change processes and access to different markets.
This article describes the revision process and the resulting publication of proposed and official updates for pharmacopoeias around the world.
An effective surveillance program for monitoring the activities of pharmacopoeias around the world requires processes, people, and tools from across a company.
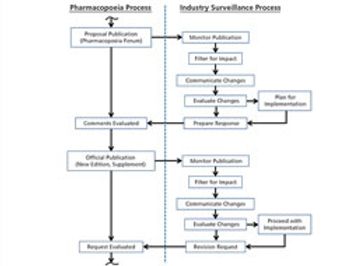
The process used to monitor and participate in pharmacopoeial changes is described.

The authors take a closer look at these ongoing efforts to harmonize compendial standards, with perspective that may be helpful in considering the future direction of pharmacopoeias.

Pharmacopoeia harmonization provides better support for global regulatory agencies and addresses the global nature of bio/pharmaceutical manufacturing and supply.

This article examines the history and evolution of the pharmacopoeias and the particular challenges that must be overcome to achieve harmonization among the pharmacopoeias.

This article provides an end-to-end compendial framework to understand why compliance with pharmacopoeia standards is challenging.
This article provides the legal and regulatory basis for pharmacopoeia compliance and illustrates pharmacopoeia impact throughout the drug product lifecycle.

In this series of articles, the authors provide an understanding about the need for pharmacopoeia compliance and practical guidance to assist those who perform this work.

In this series of articles, the authors provide an understanding about the need for pharmacopoeia compliance and practical guidance to assist those who perform this work.

In this series of articles, the authors provide an understanding about the need for pharmacopoeia compliance and practical guidance to assist those who perform this work.
Advertisement
Advertisement