




There is no harmonized guidance on pre-use integrity testing of sterilizing filters, prompting discussion among users as to whether such testing is necessary.
There is no harmonized guidance on pre-use integrity testing of sterilizing filters, prompting discussion among users as to whether such testing is necessary.

This article outlines methods, validation standards, and documentation of sterilization of single-use products using gamma irradiation.

The author looks at strategies to minimize particle levels in the finished product when using single-use technologies downstream of final capabilities.

Single-use conference roundup.

Although some aspects of single-use components can be standardized, it is unlikely that any materials or design features will become a commodity
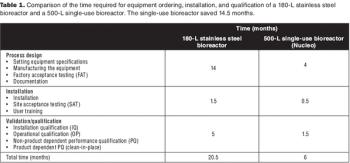
Sanofi Pasteur's disposables implementation plan is part of a larger evaluation of technology innovation. Here's how they approach it.

How a Big Pharma company tackled the move to disposable bioreactors.

How this Big Pharma company successfully implemented disposable technologies in its manufacturing plant.

Suppliers, manufacturers, and governments must work together to plan how best to develop and deploy disposable systems for emergency response.

Single-use technologies can be configured and installed fairly quickly, but are they ready to handle the urgency and scale of a pandemic?

What end users think about single-use systems.
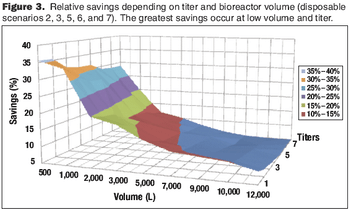
Cost modeling provides valuable insights to support strategic decision-making when implementing disposable technologies.

How to choose a disposable mixing system that fits your particular needs.
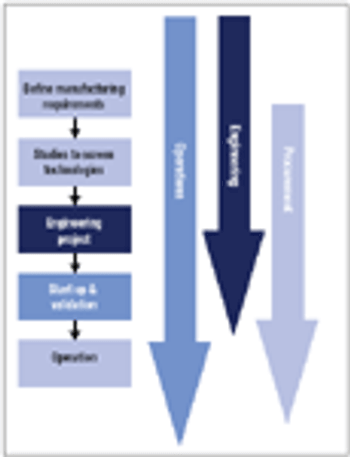
In new disposables projects, it is critical that engineering, procurement, and operations groups work together early on to manage supply chain risk.
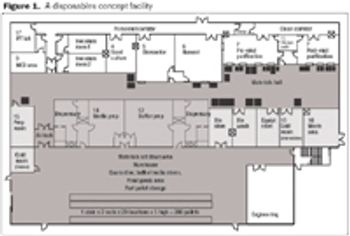
In addition to making technical developments, vendors are also looking at ways to improve supply-chain security. By offering standard, off-the-shelf products, vendors are able to shorten lead times and improve the security of supply.
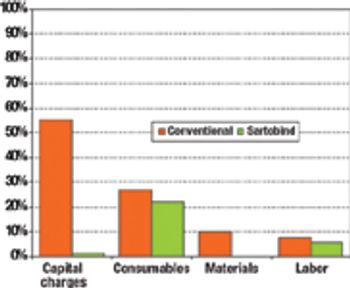
The current focus on cost-of-goods (COGS) models is underplaying the benefits of disposables technology in biopharmaceutical manufacturing. The best method for accounting for the benefits of reduced and delayed capital expenditures is through the use of NPV analysis.

Disposable technologies that mimic the conventional stainless-steel bioreactor will be most readily adopted

The use of disposables has changed significantly in the biopharmaceutical industry.