Outsourcing of manufacturing activities is expected to increase in 2019.
Outsourcing of manufacturing activities is expected to increase in 2019.

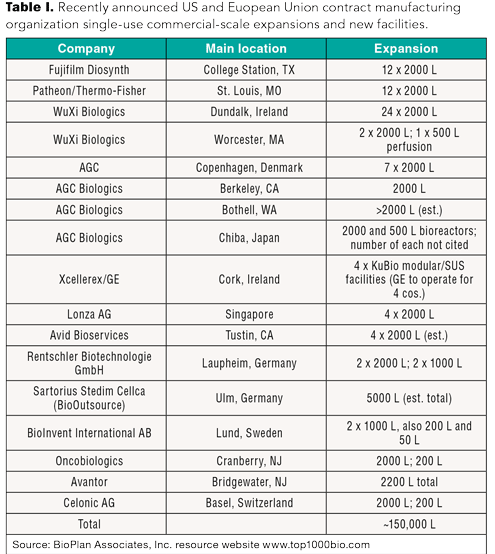
The growth in adoption of single-use systems for commercial manufacturing will be dramatic in coming years.
Partnerships, mergers, and new services indicate that biologics are continuing to influence CMOs’ and CDMOs’ decisions to expand their biopharmaceutical services.

This article highlights 15 years of changes in biopharmaceutical manufacturing.
New and expanded facilities point to the continuing growth of the biopharmaceutical industry.
CDMOs can claim credit for the robust growth of emerging bio/pharma financings.
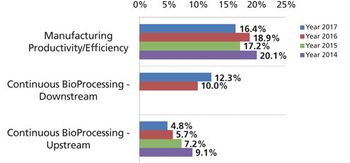
Development and adoption of new technologies create challenges that may take years to resolve.
The industry will see an impact from financing, M&As, advanced therapies, generic drugs, and the retail market in 2018.
In a productive year, 2017 was filled with acquisitions, facility expansions, and new biopharma technology.
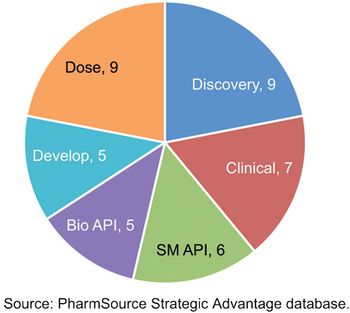
Recent acquisitions are creating CDMOs with scale that rivals global bio/pharma.
Mergers and acquisitions are positive for the CDMO industry, but there is a downside.
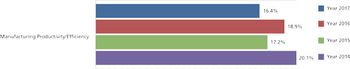
Innovation speeds discovery, drives down costs, and improves productivity.
Despite some progress, the industry is still in a wait-and-see mode regarding the administration, Congress, and FDA.
How has the bio/pharmaceutical contract manufacturing industry evolved and changed over the years and what does the future hold?
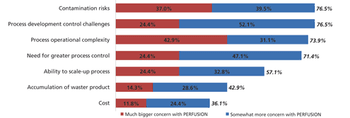
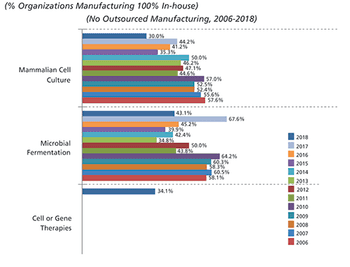
Although widespread adoption of continuous bioprocessing has been slow, some processes have been an exception.
CMOs may be gaining as strategic partners to large bio/pharma companies, but they have a much harder path to navigate.
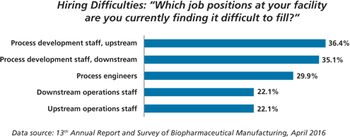
New study shows China biopharma companies face staffing shortages.
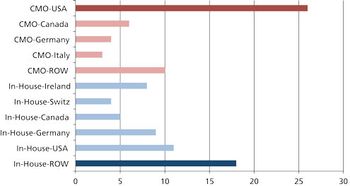
Moving global manufacturing operations may be more complicated than it appears.
The outlook for the CMO and CDMO industry may be affected by ever-changing politics.
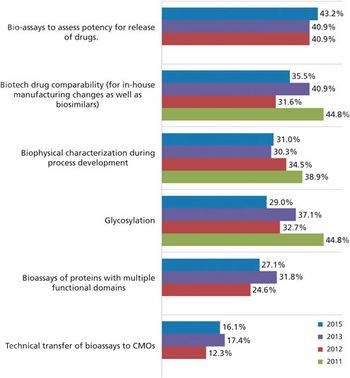
Biosimilars may be the key to CMO growth.
CMO executives are focusing on M&A activity, new business models, and fundraising limits.
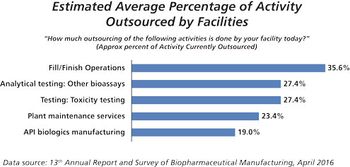
This key bioprocessing segment is expecting continued growth.
The strategies of a innovation-driven CMO may be different than a capacity-driven CMO.
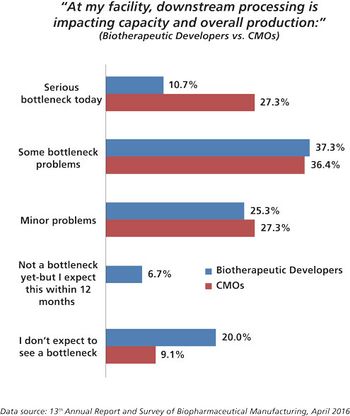
CMOs are working hard to improve performance by investigating new technologies for filtration and purification.
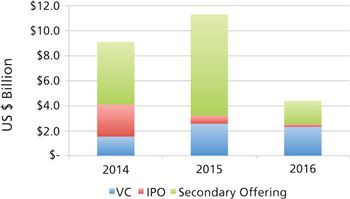
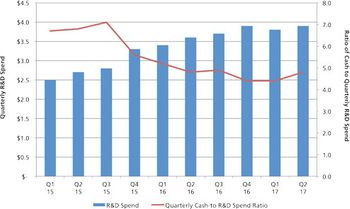
CDMOs need to be aware that unfavorable public markets put emerging bio/pharma R&D spending at risk in 2017.