




Accelerated timelines and small batch volumes of cell and gene therapies pose unique challenges for product-release testing.
Accelerated timelines and small batch volumes of cell and gene therapies pose unique challenges for product-release testing.
Both empty and filled syringes must pass a range of tests to meet quality standards for biopharmaceutical drugs.
Analytical exoglycosidases are transitioning from being largely academic tools to being suitable for glycan analytics in biopharmaceutical manufacturing.

More complex biologic samples must be evaluated to ever higher levels of specificity and sensitivity.

RGtimeline/Shutterstock.comParenteral product quality is improving.

Conducting stability testing on APIs/finished drug product helps ensure shelf-life storage.
Biopharma majors are among the industry stakeholders who have commented and raised questions about FDA’s recently proposed draft guidance for analytical assessment of similarity in biosimilars.

Process analytical testing for biopharmaceuticals requires enhanced methods due to complex bioprocesses.
A UHPLC SEC approach for protein aggregate analysis of mAbs is presented.

Increased understanding of potential impurities has spurred efforts to standardize monitoring procedures.

Time and sensitivity are essential for analytical technologies in all phases of biopharma development.
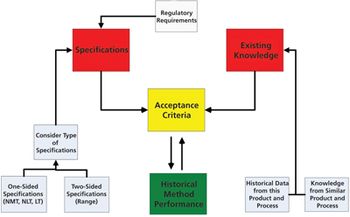
The authors present the results of a survey of biologics manufacturers to evaluate how these manufacturers transfer analytical methods.

Industry experts spoke to BioPharm International about the key considerations in the development of a drug-delivery device for a biologic drug, the importance of human factors engineering, the advantages of prefilled syringes, and the challenges in the manufacture of these devices.
USP evaluates quality attributes for synthetic peptides.