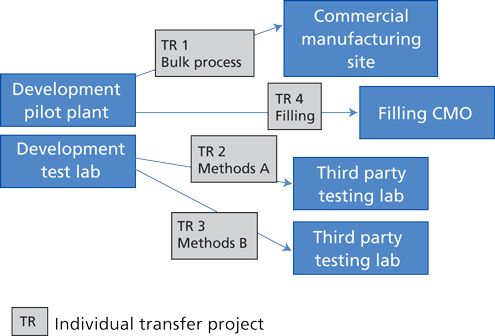

A well-constructed quality agreement can be an important tool to enable effective collaboration between owner and CMO.
A well-constructed quality agreement can be an important tool to enable effective collaboration between owner and CMO.
By embracing efficiency and quality, biopharmaceutical organizations can work better and achieve better work.
Have FDA initiatives improved manufacturing quality?

Understanding overall supplier capability versus the critical-to-quality attributes of your product can reduce both risk and cost.
Unnecessary analytical testing can lead to unnecessary costs.

The contract provider needs to know as much as the NDA holder.

How to handle and respond to a consent decree.

FDA has created a dedicated cadre of foreign drug investigators and established permanent offices worldwide.

Focusing on how risk affects the entire organization can improve the business bottom line.

Contract organizations must have highly organized teams and plans to accommodate today's audits.
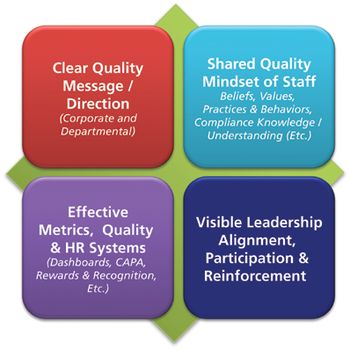
In a culture of quality, it is important that employees adopt this mindset, not because they have to, but because they understand the importance.

Why SOPs are rarely followed, often cited, and in great need of follow-through.
Incorporating regulatory requirements into the product life cycle is crucial.

A rigorous cost-benefit assessment can help to chart a cost-effective path forward.

Executive management leadership is essential in the effective implementation of QbD.

What small biotechs need to know about quality management systems.
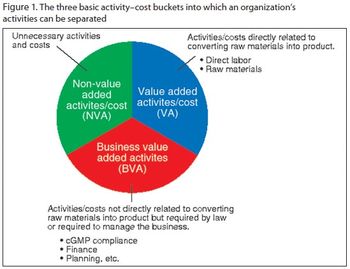
By identifying and eliminating non-value-added activities, drug manufacturers can avoid falling into the same cost-traps in the future.

The focus on the design space will lead to a new workspace, and will affect staff in the development, manufacturing, and quality functions.

A culture of quality that emphasizes business objectives, risk management, and the informed application of technology can improve compliance.

New expression systems compete for attention.

Use Lean techniques to improve manufacturing compliance

QbD can help satisfy FDA and EMEA requirements.

Every biotech company reaches a point in its development where it must decide what path it will take after it passes the start-up phase. This article discusses what the company must consider to decide what business model it will follow.
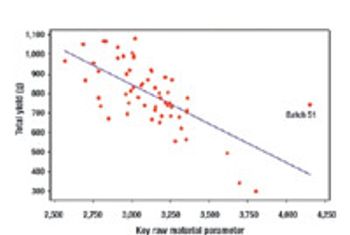
Quality by Design and Design Space can be used by companies to enhance process understanding, improve scientific rigor, and enhanced qualitative and quantative performance, as well as cost savings.
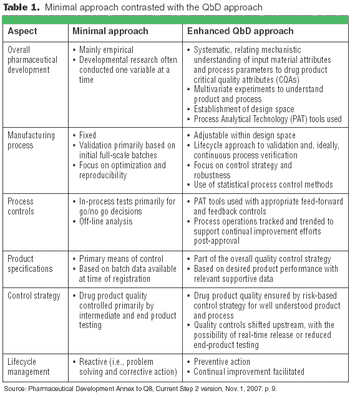
The principles of QbD can be applied to biotech development and manufacturing to help resolve many common issues. QbD scientifically provides a greater understanding of the complex relationships among product quality attributes, the manufacturing process, and clinical safety and efficacy by determining the various permutations of critical input variables that will keep the product within specification.