


Samsung will offer large-scale commercial manufacturing for drug substance and drug products to support AstraZeneca’s biologics therapeutics capabilities in the Asia-Pacific region.
Drug Substance Analysis
Latest News
Advertisement
Advertisement

Therapies for early and late treatment and passive immunization of COVID-19 are needed and can be developed using antibodies from recovered patients.

Next-generation therapeutics and regulatory requirements create demand for complex, fit-for-purpose tests.
Characterizing and controlling protein aggregation is vital to ensure safety and efficacy of a biopharmaceutical product. In this interview, important aspects of protein aggregation and the tools available to address this issue are discussed.
USP technical advisors will offer assistance to drug developers to ensure material quality and testing.

As compounds become more complex in nature and biological ingredients are more widely used, stability testing approaches must follow suit and provide flexibility for developers.

Contract service organizations can offer biopharma companies early insight into dangers that may hinder a drug’s later development.

As regulatory bodies extend the oversight of E&L testing, companies working with drug products need to make provisions on how to best comply with the evolving expectations.
Through the agreement, Samsung will provide flexible business terms while offering full regulatory support and improved batch release from small to large scale.
Particulates or aggregates are a notable challenge for injectables, but there are several methods available to help with identification during formulation and development.

Stability testing for biologics is more complex than for small-molecule drugs, so companies should be aware of the potentially serious issues that can be costly and jeopardize drug development.
FDA and ICH seek comment on new exposure levels for cadmium in drug products.
Transcriptomics plays a role in influencing the production of recombinant therapeutics in microbial and mammalian hosts.

Conducting stability testing on APIs/finished drug product helps ensure shelf-life storage.
A case study demonstrates that affinity chromatography can offer efficiency and scalability for gene therapy manufacturing using viral vectors.
This article summarizes the approaches, challenges, and future perspectives for the characterization of N-glycans in biopharmaceutical products.

Increased understanding of potential impurities has spurred efforts to standardize monitoring procedures.
An increase in biologics raises awareness of particle generation and its role in negative patient outcomes.

The complex nature of biologics adds additional CQAs that must be determined to ensure the safe development of biologics
The objective of this study was to assess the impact of manufacturing-scale, freeze-thaw conditions on aggregation and subvisible particle formation of a monoclonal antibody solution (mAb-A; IgG1) using a small-scale model.

This study aims at understanding the differences between porcine and bovine trypsin from both pancreatic and recombinant origins.
The development of mAb formulations poses challenges at the manufacturing, stability, analytical, and administration levels.
Interactions between biologic drug products and the components of prefilled syringes can cause protein aggregation, but there are alternative materials that can help mitigate this problem.
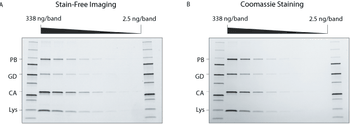
Innovations in electrophoresis and chromatography upstream of protein characterization can accelerate research.
Multiple methods are required for detecting and removing protein impurities.
Advertisement
Advertisement