



Compendial Compliance
Latest News
Advertisement
Latest Videos
Advertisement
More News
Maintaining compliance to compendial requirements should be straightforward, says Susan J. Schniepp, distinguished fellow at Regulatory Compliance Associates.

This article focuses on drawing parallels between ICH Q14/Q2(R2), United States Pharmacopeia (USP) <1220>, and International Organization for Standardization/International Electrotechnical Commission (ISO/IEC) 17025:2017.

This article provides an overview of validation concept principles evolution to a life cycle risk-based approach with focus on compendial perspectives.

Awareness of recently implemented—or ongoing—advances by the pharmacopoeias can help biotherapeutic manufacturers remain compliant with current requirements.
Experts Susan J. Schniepp, distinguished fellow for Regulatory Compliance Associates, and Steven J. Lynn, executive vice-president, Pharmaceuticals for Regulatory Compliance Associates, discuss the verification of compendial methods.
USP is developing mRNA quality guidelines to help companies and regulators bring innovative medicines to market faster.

BioPharm International checked in with AAPS, IPEC-Americas, and PDA to get an update on how the organizations are navigating the pandemic and planning for the future.

Public health challenges have highlighted the need for agility in maintaining the quality of medicines.


The European Pharmacopoeia is preparing for 2021 while also supporting the industry to develop vaccines and treatments for COVID-19.
The guidelines are part of the independent control of pandemic COVID-19 vaccine batches.

Processes, people, and tools are necessary to comply with the pharmacopoeia and approved drug product registrations.
The Committee for Human Medicinal Products (CHMP) of the European Medicines Agency (EMA) has issued its final opinion on measures for companies to take that will limit the presence of nitrosamines in human medicines.
The ASTM standard describes how to evaluate single-use systems for foreign particle analysis.
USP technical advisors will offer assistance to drug developers to ensure material quality and testing.

This article details the more operational aspects of monograph submissions, answering the question of how to participate.
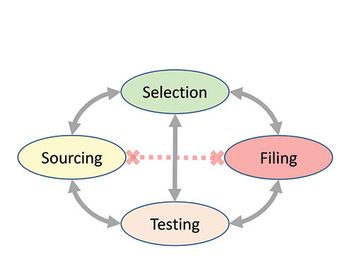
The authors present a case study with raw materials and excipients, where a consistent, cross-functional approach is needed to ensure the appropriate selection, sourcing, testing, and filing of the materials used to manufacture bio/pharmaceutical products in a global environment, ensuring compliance with applicable compendial and regulatory requirements.

This series is intended to address the challenges for the industry to comply with pharmacopoeial requirements. This article returns to this important topic with a case study at the intersection of monograph development and compliance.

This article summarizes all the considerations that go into a company’s compendial affairs program and to look ahead at topics that will likely result in further evolution in the pharmacopoeias around the world. This look into what is on the horizon is important to help companies prepare for the inevitable changes and ensure the continued supply of quality medicines to patients globally.

Monographs are developed based on the submission of information and materials from a company having regulatory approval for the product, and this submission feeds into the pharmacopoeia revision process.

This final article in the series has two purposes: to summarize all the considerations that go into a company’s compendial affairs program and to look ahead at topics that will likely result in further evolution in the pharmacopoeias around the world.

This article returns to the topic of complying with pharmacopoeial requirements with a case study at the intersection of monograph development and compliance.
An evaluation by USP indicates bovine heparin is a potential alternative to porcine heparin.

The revision process and the resulting publication of proposed and official updates for pharmacopoeias around the world are described.

This article describes the revision process and the resulting publication of proposed and official updates for pharmacopoeias around the world.
Advertisement
Advertisement