




Viral clearance processes and guidance must evolve along with newer biotherapeutic modalities.
Viral clearance processes and guidance must evolve along with newer biotherapeutic modalities.
The second half of this article describes the testing methods used by the authors to demonstrate the applicability of single-use mixing technology for virus inactivation.
This article explores the use of single-use mixing technology in a detergent-based virus inactivation step during a monoclonal antibody production process.
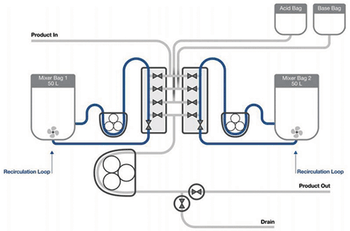
Testing demonstrates an automated semi-continuous process strategy for viral inactivation with steps that mimic batch processing.

Viral vaccines and viral vectors used in biotherapeutic applications carry the risk of microbial contamination, which must be addressed.
The new resin used a combination of “jetting” technology and a high-performance Protein A ligand.
Process understanding and careful assessment of risks are essential in developing viral clearance programs.
Detecting viral contaminants in biologic-based medicines-and identifying their source-requires a holistic testing approach.

This study outlines methods for an alternative protein-polishing process for challenging proteins.
A case study demonstrates that affinity chromatography can offer efficiency and scalability for gene therapy manufacturing using viral vectors.

Protecting against microbiological contaminationover the whole manufacturing process grows increasingly important.

Continuous processing of 100 g of monoclonal antibody in 24 hours has been demonstrated using lab-scale equipment.

Increased understanding of potential impurities has spurred efforts to standardize monitoring procedures.

The unique structures of fusion proteins lead to expression, heterogeneity, and stability issues.
For cellular materials, new ultra scale-down devices inform large-scale downstream processing techniques.

A novel coiled flow inversion reactor (CFIR) improves process productivity and performance.

A new virus-retentive membrane may be used to filter chemical-defined cell culture media for risk mitigation.

Advances in cell culture media technology have helped achieve safer biologics.

Pressures to accelerate current and next-gen therapies are challenging traditional microbiological testing methods.

Advances in cell line engineering, process optimization, and in-vitro glycosylation are making a difference.
Testing product and process intermediates alone is helpful, but does not provide a complete solution to viral safety. This article proposes integrated solutions for systemic and proactive viral risk mitigation.
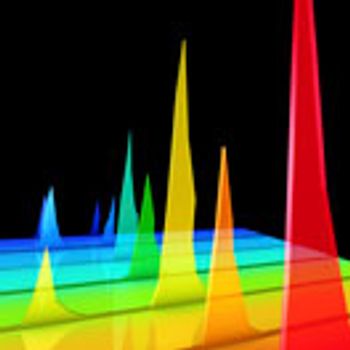
The authors provide application data to support the use of SEC beyond small-scale operations.

BioPharm International sat down with Kevin Isett, PhD, co-founder and CEO of Avitide, to find out why he thinks the company’s tailored approach to purification resins will change the face of biopharmaceutical separation.
Whether taking an upstream, downstream or holistic approach, there are many factors to consider when choosing viral clearance methods.

Industry experts discuss the development of process chromatography in bioprocessing.